O. Baraka , M. Fodil , A. Mokadem , Mohammed Benali Kanoun , Souraya Goumri-Said
{"title":"A Comparative analysis of the physical properties of predicted MAX phases V2SnN and V2SnB with the synthesized V2SnC: Insights from DFT calculations","authors":"O. Baraka , M. Fodil , A. Mokadem , Mohammed Benali Kanoun , Souraya Goumri-Said","doi":"10.1016/j.comptc.2024.114971","DOIUrl":null,"url":null,"abstract":"<div><div>The M<sub>n+1</sub>AX<sub>n</sub> phases, known for their unique combination of metallic and ceramic properties, have attracted significant attention due to their potential applications in extreme environments. In this study, we use density functional theory to investigate the structural, electronic, elastic, mechanical, and thermodynamic properties of V<sub>2</sub>SnX (X = B and N) compounds. Our results confirm that both compounds are elastically, thermodynamically, and dynamically stable, with a ductile nature. The metallic behavior is further supported by electronic band structures and density of states analysis, with V-<em>d</em> states playing a key role in electrical conductivity. Using the quasi-harmonic Debye model, we also examine the effects of temperature and pressure, finding that the bulk modulus and Debye temperature decrease with rising pressure below 50 GPa. Based on these findings, V<sub>2</sub>SnX compounds are ideal candidates for high-performance applications in harsh conditions, where materials need to maintain stability, conductivity, and resistance to thermal and mechanical stress.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114971"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24005103","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/3 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
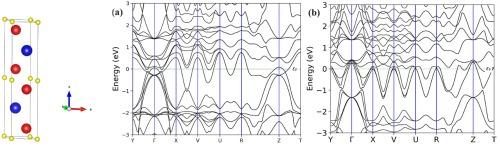
The Mn+1AXn phases, known for their unique combination of metallic and ceramic properties, have attracted significant attention due to their potential applications in extreme environments. In this study, we use density functional theory to investigate the structural, electronic, elastic, mechanical, and thermodynamic properties of V2SnX (X = B and N) compounds. Our results confirm that both compounds are elastically, thermodynamically, and dynamically stable, with a ductile nature. The metallic behavior is further supported by electronic band structures and density of states analysis, with V-d states playing a key role in electrical conductivity. Using the quasi-harmonic Debye model, we also examine the effects of temperature and pressure, finding that the bulk modulus and Debye temperature decrease with rising pressure below 50 GPa. Based on these findings, V2SnX compounds are ideal candidates for high-performance applications in harsh conditions, where materials need to maintain stability, conductivity, and resistance to thermal and mechanical stress.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
