{"title":"Modeling of FAK-PROTAC candidates from GSK2256098 analogs for targeted protein degradation","authors":"Vikas Kumar , Shraddha Parate , Hyeon-Su Ro , Tae Sung Jung , Keun Woo Lee","doi":"10.1016/j.bbrc.2024.151001","DOIUrl":null,"url":null,"abstract":"<div><div>Protein inhibition via the traditional drug-designing approach has been shown to be an effective method for developing numerous small-molecule-based therapeutics. In the last decade, small inhibitors-guided protein degradation has arisen as an alternative method with the potential to fulfill the drug requirement for undruggable targets. Focal adhesion kinase (FAK) is a crucial modulator of the growth and spread of tumors, apart from it also acts as a scaffold for signaling of other proteins. FAK inhibitors have thus far had unsatisfactory results in clinical trials for cancer applications. Unlike prior attempts to control FAK expression, which were restricted to kinase domain inhibition with limited success in clinical research, protein degradation has the potential to concurrently disrupt FAK's kinase and scaffolding function. Recently, several FAK degraders were reported based on FAK Type I inhibitors using complex chemical synthesis approaches. Interestingly, recently a ternary complex was published revealing the binding mode of the FAK-PROTAC-E3 complex. This complex opens an avenue for the development of rational PROTAC design against FAK protein. Therefore, in the present study, we selected the most active Type I FAK inhibitor GSK2256098. The binding mode of the inhibitor prompted us to identify the most suitable analog for PROTAC design. We have identified a high-affinity analog that is suitable for PTOTAC design through the application of molecular docking (MD) and molecular dynamics simulations (MDS). Further based on the ternary FAK-PROTAC-E3 complex we build a binary complex FAK-Hit-E3-VHL between both proteins. Using the structure-based approach ten different potential FAK PROTACs candidates were designed. The stability of the complexes was analyzed using MDS and binding free energies were used to predict the binding affinity. Finally, based on desirable intermolecular interactions with the target and E3 ligase ProTAC4 was selected as the best candidate when compared with known FAK PROTAC GSK215.</div></div>","PeriodicalId":8779,"journal":{"name":"Biochemical and biophysical research communications","volume":"740 ","pages":"Article 151001"},"PeriodicalIF":2.2000,"publicationDate":"2024-12-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochemical and biophysical research communications","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0006291X24015377","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/17 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
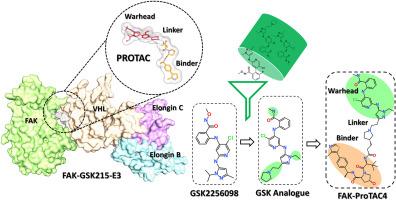
Protein inhibition via the traditional drug-designing approach has been shown to be an effective method for developing numerous small-molecule-based therapeutics. In the last decade, small inhibitors-guided protein degradation has arisen as an alternative method with the potential to fulfill the drug requirement for undruggable targets. Focal adhesion kinase (FAK) is a crucial modulator of the growth and spread of tumors, apart from it also acts as a scaffold for signaling of other proteins. FAK inhibitors have thus far had unsatisfactory results in clinical trials for cancer applications. Unlike prior attempts to control FAK expression, which were restricted to kinase domain inhibition with limited success in clinical research, protein degradation has the potential to concurrently disrupt FAK's kinase and scaffolding function. Recently, several FAK degraders were reported based on FAK Type I inhibitors using complex chemical synthesis approaches. Interestingly, recently a ternary complex was published revealing the binding mode of the FAK-PROTAC-E3 complex. This complex opens an avenue for the development of rational PROTAC design against FAK protein. Therefore, in the present study, we selected the most active Type I FAK inhibitor GSK2256098. The binding mode of the inhibitor prompted us to identify the most suitable analog for PROTAC design. We have identified a high-affinity analog that is suitable for PTOTAC design through the application of molecular docking (MD) and molecular dynamics simulations (MDS). Further based on the ternary FAK-PROTAC-E3 complex we build a binary complex FAK-Hit-E3-VHL between both proteins. Using the structure-based approach ten different potential FAK PROTACs candidates were designed. The stability of the complexes was analyzed using MDS and binding free energies were used to predict the binding affinity. Finally, based on desirable intermolecular interactions with the target and E3 ligase ProTAC4 was selected as the best candidate when compared with known FAK PROTAC GSK215.
期刊介绍:
Biochemical and Biophysical Research Communications is the premier international journal devoted to the very rapid dissemination of timely and significant experimental results in diverse fields of biological research. The development of the "Breakthroughs and Views" section brings the minireview format to the journal, and issues often contain collections of special interest manuscripts. BBRC is published weekly (52 issues/year).Research Areas now include: Biochemistry; biophysics; cell biology; developmental biology; immunology
; molecular biology; neurobiology; plant biology and proteomics



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
