{"title":"Modulated electronic properties of borophene nanoribbons using copper and oxygen atoms","authors":"Weihua Wang, Junchi Ma, Yuxuan Wang, Fushi Jiang, Kunpeng Zhou, Peifang Li","doi":"10.1016/j.chemphys.2024.112520","DOIUrl":null,"url":null,"abstract":"<div><div>The electronic and optical properties of borophene nanoribbons, influenced by the adsorption of copper (Cu) and oxygen (O) atoms in various configurations, were studied using first-principles calculations. The findings indicated that the most stable configuration, with the minimum adsorption energy of −9.09 eV, was achieved with double-upper center adsorbed double O atoms. By varying the adsorption positions and the number of Cu and O atoms, borophene nanoribbons were successfully tuned to transform into direct and indirect band-gap semiconductors. In particular, borophene nanoribbons with single and double-upper center adsorbed Cu atoms exhibited direct band gaps of 0.083 eV and 0.185 eV, respectively. In contrast, the double-upper center adsorbed double O atoms configuration resulted in an indirect band gap of 0.039 eV. The results further demonstrated that the Fermi levels crossed into the valence band, displaying p-type degenerate semiconductor characteristics for configurations with single and double-upper center adsorbed Cu atoms as well as double-upper center adsorbed double O atoms.</div></div>","PeriodicalId":272,"journal":{"name":"Chemical Physics","volume":"590 ","pages":"Article 112520"},"PeriodicalIF":2.4000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301010424003495","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/17 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
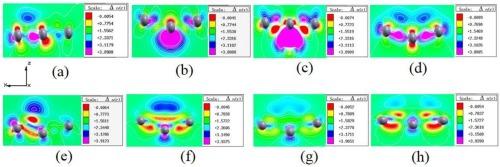
The electronic and optical properties of borophene nanoribbons, influenced by the adsorption of copper (Cu) and oxygen (O) atoms in various configurations, were studied using first-principles calculations. The findings indicated that the most stable configuration, with the minimum adsorption energy of −9.09 eV, was achieved with double-upper center adsorbed double O atoms. By varying the adsorption positions and the number of Cu and O atoms, borophene nanoribbons were successfully tuned to transform into direct and indirect band-gap semiconductors. In particular, borophene nanoribbons with single and double-upper center adsorbed Cu atoms exhibited direct band gaps of 0.083 eV and 0.185 eV, respectively. In contrast, the double-upper center adsorbed double O atoms configuration resulted in an indirect band gap of 0.039 eV. The results further demonstrated that the Fermi levels crossed into the valence band, displaying p-type degenerate semiconductor characteristics for configurations with single and double-upper center adsorbed Cu atoms as well as double-upper center adsorbed double O atoms.
通过第一原理计算,研究了硼吩纳米带的电子和光学特性受铜(Cu)和氧(O)原子以不同构型吸附的影响。结果表明,双上中心吸附双 O 原子的构型最稳定,吸附能最小,为 -9.09 eV。通过改变 Cu 原子和 O 原子的吸附位置和数量,成功地将硼吩纳米带调整为直接和间接带隙半导体。其中,单中心和双上中心吸附 Cu 原子的硼吩纳米带的直接带隙分别为 0.083 eV 和 0.185 eV。相比之下,双上中心吸附双 O 原子构型的间接带隙为 0.039 eV。结果进一步表明,费米级跨入价带,在单中心和双上中心吸附铜原子以及双上中心吸附双 O 原子的配置中显示出 p 型退化半导体特性。
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
