Dongchen Chu , CuiCui Ji , Yu Zhang , Chaochun Wei , Xiaokun Zhang , Qidi Zhong , Hong Yan , Juan Wang
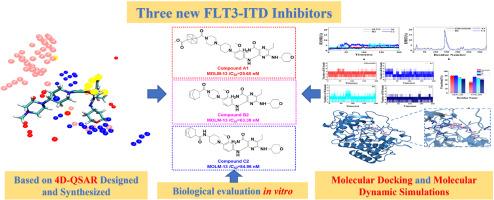
{"title":"Identification of inhibitors targeting the FLT3-ITD mutation through 4D-QSAR, in vitro, and in silico","authors":"Dongchen Chu , CuiCui Ji , Yu Zhang , Chaochun Wei , Xiaokun Zhang , Qidi Zhong , Hong Yan , Juan Wang","doi":"10.1016/j.ejmech.2024.117089","DOIUrl":null,"url":null,"abstract":"<div><div>The FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) mutation is a key target for acute myeloid leukemia (AML) treatment. The second-generation inhibitors such as Gilteritinib still present off-target effects and associated side effects. Therefore, identifying novel FLT3-ITD inhibitors has become a promising strategy for AML treatment. In this study, a 4D-QSAR model was developed based on Gilteritinib and its analogues, and it was found that introducing hydrophobic bulky groups at the piperazine or piperidine of Gilteritinib would enhance the binding affinity to FLT3-ITD. So, three series of targeted compounds (<strong>A1</strong>-<strong>A5</strong>, <strong>B1</strong>–<strong>B5</strong> and <strong>C1</strong>–<strong>C5</strong>) were designed and synthesized. The antiproliferative activity against MOLM-13 cells was evaluated <em>in vitro</em>. Compound <strong>A1</strong> (<em>IC</em><sub><em>50</em></sub> = 25.65 nM), with a cubane group at the piperazine position; Compounds <strong>B2</strong> (<em>IC</em><sub><em>50</em></sub> = 63.38 nM) and <strong>C2</strong> (<em>IC</em><sub><em>50</em></sub> = 54.96 nM), with a norbornene group at the piperidine position, showed the strongest inhibition in their series. Their <em>IC</em><sub><em>50</em></sub> values were comparable to that of the positive control Gilteritinib (<em>IC</em><sub><em>50</em></sub> = 22.37 nM). FLT3-ITD was confirmed as the degradation target through a kinase inhibition assay, where the <em>IC</em><sub><em>50</em></sub> values were 2.12 nM (Compound <strong>A1</strong>), 1.29 nM (Compound <strong>B2</strong>), and 3.06 nM (Compound <strong>C2</strong>), which were comparable to that of Gilteritinib (<em>IC</em><sub><em>50</em></sub> = 0.43 nM). Additionally, molecular docking and molecular dynamics (MD) simulations showed that Compounds <strong>A1</strong>, <strong>B2</strong>, and <strong>C2</strong> had similar binding modes to that of Gilteritinib with more stable affinities. Overall, these results demonstrated that Compounds <strong>A1</strong>, <strong>B2</strong>, and <strong>C2</strong> were promising inhibitors for targeting AML with FLT3-ITD mutation.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"282 ","pages":"Article 117089"},"PeriodicalIF":5.9000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523424009711","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/26 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
The FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) mutation is a key target for acute myeloid leukemia (AML) treatment. The second-generation inhibitors such as Gilteritinib still present off-target effects and associated side effects. Therefore, identifying novel FLT3-ITD inhibitors has become a promising strategy for AML treatment. In this study, a 4D-QSAR model was developed based on Gilteritinib and its analogues, and it was found that introducing hydrophobic bulky groups at the piperazine or piperidine of Gilteritinib would enhance the binding affinity to FLT3-ITD. So, three series of targeted compounds (A1-A5, B1–B5 and C1–C5) were designed and synthesized. The antiproliferative activity against MOLM-13 cells was evaluated in vitro. Compound A1 (IC50 = 25.65 nM), with a cubane group at the piperazine position; Compounds B2 (IC50 = 63.38 nM) and C2 (IC50 = 54.96 nM), with a norbornene group at the piperidine position, showed the strongest inhibition in their series. Their IC50 values were comparable to that of the positive control Gilteritinib (IC50 = 22.37 nM). FLT3-ITD was confirmed as the degradation target through a kinase inhibition assay, where the IC50 values were 2.12 nM (Compound A1), 1.29 nM (Compound B2), and 3.06 nM (Compound C2), which were comparable to that of Gilteritinib (IC50 = 0.43 nM). Additionally, molecular docking and molecular dynamics (MD) simulations showed that Compounds A1, B2, and C2 had similar binding modes to that of Gilteritinib with more stable affinities. Overall, these results demonstrated that Compounds A1, B2, and C2 were promising inhibitors for targeting AML with FLT3-ITD mutation.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
