Carlos V. Santos-Jr, Elfi Kraka, Renaldo T. Moura Jr
{"title":"Chemical Bond Overlap Descriptors From Multiconfiguration Wavefunctions","authors":"Carlos V. Santos-Jr, Elfi Kraka, Renaldo T. Moura Jr","doi":"10.1002/jcc.27534","DOIUrl":null,"url":null,"abstract":"<p>The chemical bond is a fundamental concept in chemistry, and various models and descriptors have evolved since the advent of quantum mechanics. This study extends the overlap density and its topological descriptors (OP/TOP) to multiconfigurational wavefunctions. We discuss a comparative analysis of OP/TOP descriptors using CASSCF and DCD-CAS(2) wavefunctions for a diverse range of molecular systems, including X–O bonds in X–OH (XH, Li, Na, H<sub>2</sub>B, H<sub>3</sub>C, H<sub>2</sub>N, HO, F) and Li–X′ (XF, Cl, and Br). Results show that OP/TOP aligns with bonding models like the quantum theory of atoms in molecules (QTAIM) and local vibrational modes theory, revealing insights such as overlap densities shifting towards the more electronegative atom in polar bonds. The Li–F dissociation profile using OP/TOP descriptors demonstrated sensitivity to ionic/neutral inversion during Li–F dissociation, highlighting their potential for elucidating complex bond phenomena and offering new avenues for understanding multiconfigurational chemical bond dynamics.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-11-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27534","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27534","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
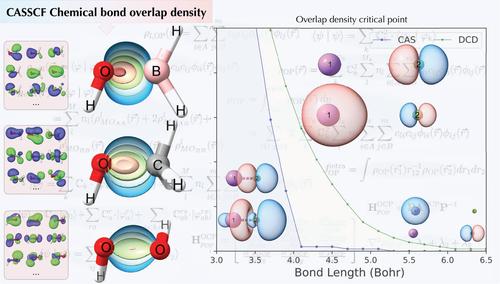
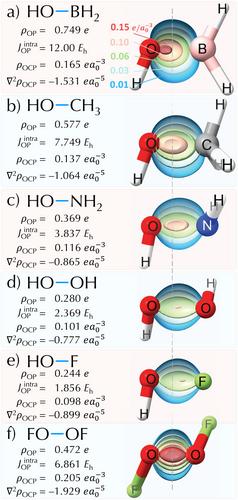
The chemical bond is a fundamental concept in chemistry, and various models and descriptors have evolved since the advent of quantum mechanics. This study extends the overlap density and its topological descriptors (OP/TOP) to multiconfigurational wavefunctions. We discuss a comparative analysis of OP/TOP descriptors using CASSCF and DCD-CAS(2) wavefunctions for a diverse range of molecular systems, including X–O bonds in X–OH (XH, Li, Na, H2B, H3C, H2N, HO, F) and Li–X′ (XF, Cl, and Br). Results show that OP/TOP aligns with bonding models like the quantum theory of atoms in molecules (QTAIM) and local vibrational modes theory, revealing insights such as overlap densities shifting towards the more electronegative atom in polar bonds. The Li–F dissociation profile using OP/TOP descriptors demonstrated sensitivity to ionic/neutral inversion during Li–F dissociation, highlighting their potential for elucidating complex bond phenomena and offering new avenues for understanding multiconfigurational chemical bond dynamics.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
