Li Ping Ding , Fei Yue Qiao , Hong Yuan Xu , Shao Fei Lei , Peng Shao
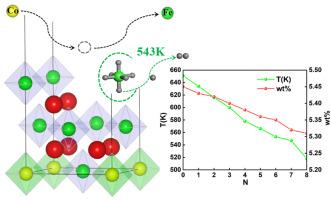
{"title":"Investigation on the effect of Co doping on structure, electronic, and hydrogen storage properties of Mg2FeH6","authors":"Li Ping Ding , Fei Yue Qiao , Hong Yuan Xu , Shao Fei Lei , Peng Shao","doi":"10.1016/j.jmgm.2024.108916","DOIUrl":null,"url":null,"abstract":"<div><div>Metal hydrides, particularly magnesium-based materials, exhibit excellent hydrogen storage capabilities. Among these, Mg<sub>2</sub>FeH<sub>6</sub> stands out for its high hydrogen storage capacity, but it faces limitations due to low thermodynamic stability and high hydrogen desorption temperature. To overcome these challenges, we investigated the potential of Co doping to improve hydrogen storage properties. Based on first-principles calculations, we systematically explored the structures, electronic properties and hydrogen storage capabilities of a novel Mg-Fe-Co-H alloy. We found that Co doping significantly reduced the band gap by 1.14 eV, promoting electron transitions and accelerating hydrogen desorption kinetics. Additionally, Co doping alters the Fe-H interaction, increasing bond lengths and facilitating the hydrogen dissociation. Although Co doping slightly decreases hydrogen storage capability of Mg<sub>2</sub>FeH<sub>6</sub> (from 5.45 <em>wt</em>% to 5.32 <em>w</em>t%), it significantly lowers desorption temperature from 651 K (Mg<sub>2</sub>FeH<sub>6</sub>) to 543 K (Mg<sub>2</sub>Fe<sub>1/8</sub>Co<sub>7/8</sub>H<sub>6</sub>). This study highlights the innovative potential of Co doping to enhance the performance of Mg<sub>2</sub>FeH<sub>6</sub>-based hydrogen storage materials, offering promising prospects for advancing hydrogen energy technologies.</div></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"135 ","pages":"Article 108916"},"PeriodicalIF":3.0000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S109332632400216X","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Metal hydrides, particularly magnesium-based materials, exhibit excellent hydrogen storage capabilities. Among these, Mg2FeH6 stands out for its high hydrogen storage capacity, but it faces limitations due to low thermodynamic stability and high hydrogen desorption temperature. To overcome these challenges, we investigated the potential of Co doping to improve hydrogen storage properties. Based on first-principles calculations, we systematically explored the structures, electronic properties and hydrogen storage capabilities of a novel Mg-Fe-Co-H alloy. We found that Co doping significantly reduced the band gap by 1.14 eV, promoting electron transitions and accelerating hydrogen desorption kinetics. Additionally, Co doping alters the Fe-H interaction, increasing bond lengths and facilitating the hydrogen dissociation. Although Co doping slightly decreases hydrogen storage capability of Mg2FeH6 (from 5.45 wt% to 5.32 wt%), it significantly lowers desorption temperature from 651 K (Mg2FeH6) to 543 K (Mg2Fe1/8Co7/8H6). This study highlights the innovative potential of Co doping to enhance the performance of Mg2FeH6-based hydrogen storage materials, offering promising prospects for advancing hydrogen energy technologies.
金属氢化物,特别是镁基材料,表现出优异的储氢能力。其中,Mg2FeH6具有较高的储氢能力,但热力学稳定性低,脱氢温度高,限制了Mg2FeH6的应用。为了克服这些挑战,我们研究了Co掺杂提高储氢性能的潜力。基于第一性原理计算,我们系统地探索了一种新型Mg-Fe-Co-H合金的结构、电子性能和储氢能力。我们发现,Co掺杂显著降低了1.14 eV的带隙,促进了电子跃迁,加速了氢的脱附动力学。此外,Co掺杂改变了Fe-H相互作用,增加了键长,促进了氢的解离。虽然Co掺杂略微降低了Mg2FeH6的储氢能力(从5.45 wt%降至5.32 wt%),但显著降低了解吸温度,从651 K (Mg2FeH6)降至543 K (Mg2Fe1/8Co7/8H6)。该研究强调了Co掺杂提高mg2feh6基储氢材料性能的创新潜力,为推进氢能技术提供了广阔的前景。
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
