Workflow for practical quantum chemical calculations with a quantum phase estimation algorithm: electronic ground and π–π* excited states of benzene and its derivatives†
{"title":"Workflow for practical quantum chemical calculations with a quantum phase estimation algorithm: electronic ground and π–π* excited states of benzene and its derivatives†","authors":"Yusuke Ino, Misaki Yonekawa, Hideto Yuzawa, Yuichiro Minato and Kenji Sugisaki","doi":"10.1039/D4CP03454F","DOIUrl":null,"url":null,"abstract":"<p >Quantum computers are expected to perform full-configuration interaction calculations with less computational resources compared to classical ones, thanks to the use of quantum phase estimation (QPE) algorithms. However, only a limited number of QPE-based quantum chemical calculations have been reported even for numerical simulations on a classical computer, and the practical workflow for the QPE computation has not yet been established. In this paper, we report the QPE simulations of the electronic ground and the π–π* excited singlet state of benzene and its chloro- and nitro-derivatives as the representative industrially important systems, with the aid of GPGPU acceleration of quantum circuit simulations. We adopted the pseudo-natural orbitals obtained from the MP2 calculation as the basis for the wave function expansion, the CISD calculation within the active space to find the main electronic configurations to be included in the input wave function of the excited state, and the technique to reduce the truncation error of the calculated total energies. The proposed computational workflow is easily applicable to other molecules and can be a standard approach for performing QPE-based quantum chemical calculations of practical molecules.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 48","pages":" 30044-30054"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp03454f?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp03454f","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
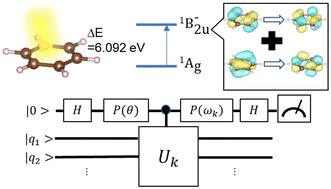
Quantum computers are expected to perform full-configuration interaction calculations with less computational resources compared to classical ones, thanks to the use of quantum phase estimation (QPE) algorithms. However, only a limited number of QPE-based quantum chemical calculations have been reported even for numerical simulations on a classical computer, and the practical workflow for the QPE computation has not yet been established. In this paper, we report the QPE simulations of the electronic ground and the π–π* excited singlet state of benzene and its chloro- and nitro-derivatives as the representative industrially important systems, with the aid of GPGPU acceleration of quantum circuit simulations. We adopted the pseudo-natural orbitals obtained from the MP2 calculation as the basis for the wave function expansion, the CISD calculation within the active space to find the main electronic configurations to be included in the input wave function of the excited state, and the technique to reduce the truncation error of the calculated total energies. The proposed computational workflow is easily applicable to other molecules and can be a standard approach for performing QPE-based quantum chemical calculations of practical molecules.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
