Abu Salim Mustafa, Mohd Wasif Khan, Nazima Habibi, Wadha Alfouzan
{"title":"Whole-Genome Sequencing of Brucella melitensis Isolates from Kuwait for the Identification of Biovars, Variants, and Relationship within a Biovar.","authors":"Abu Salim Mustafa, Mohd Wasif Khan, Nazima Habibi, Wadha Alfouzan","doi":"10.1159/000542867","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.</p><p><strong>Methods: </strong>DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using \"bwa-mem\" and SAMtools/VCFtools, respectively.</p><p><strong>Results: </strong>The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.</p><p><strong>Conclusions: </strong>Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.</p><p><strong>Objective: </strong>The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.</p><p><strong>Methods: </strong>DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using \"bwa-mem\" and SAMtools/VCFtools, respectively.</p><p><strong>Results: </strong>The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.</p><p><strong>Conclusions: </strong>Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.</p>","PeriodicalId":18455,"journal":{"name":"Medical Principles and Practice","volume":" ","pages":"152-161"},"PeriodicalIF":2.2000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11936451/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medical Principles and Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1159/000542867","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/29 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
Objective: The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.
Methods: DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using "bwa-mem" and SAMtools/VCFtools, respectively.
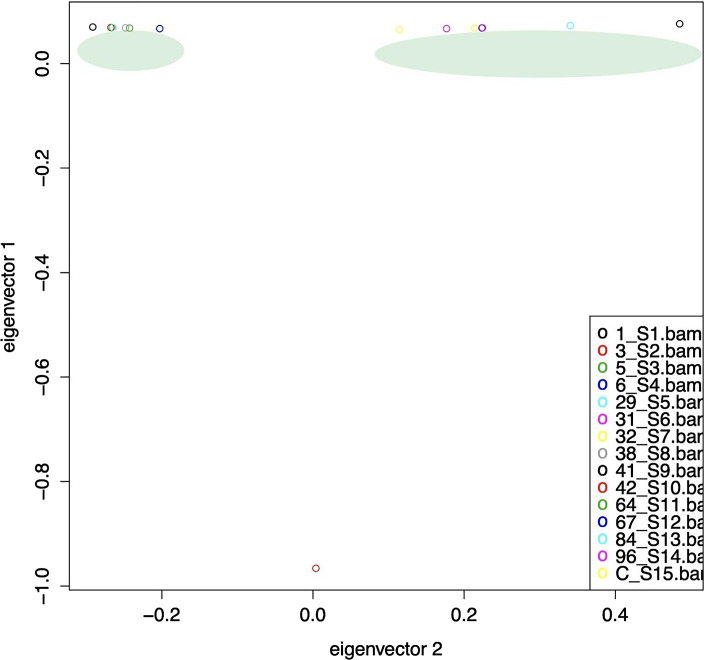
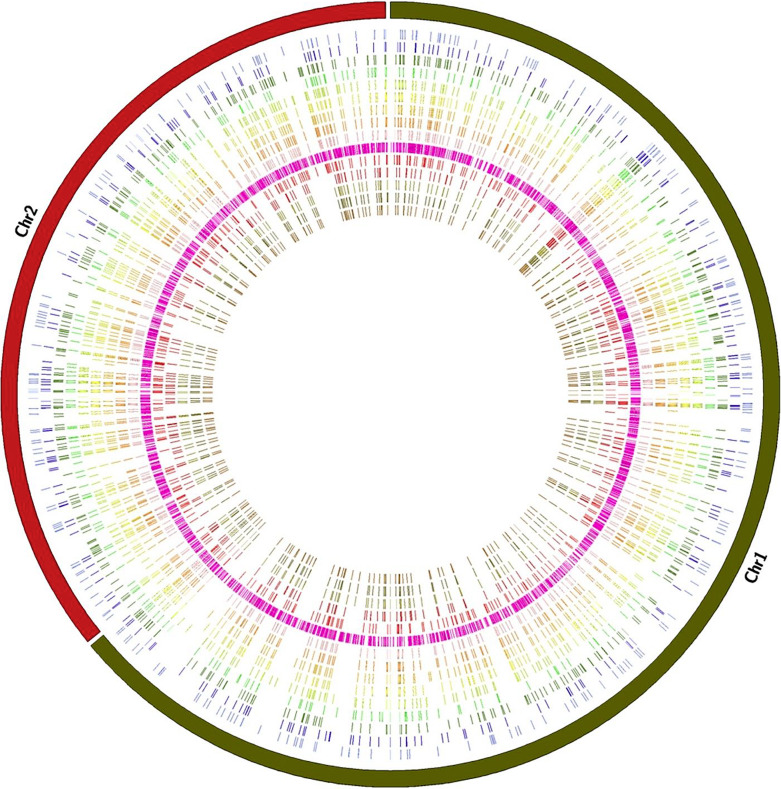
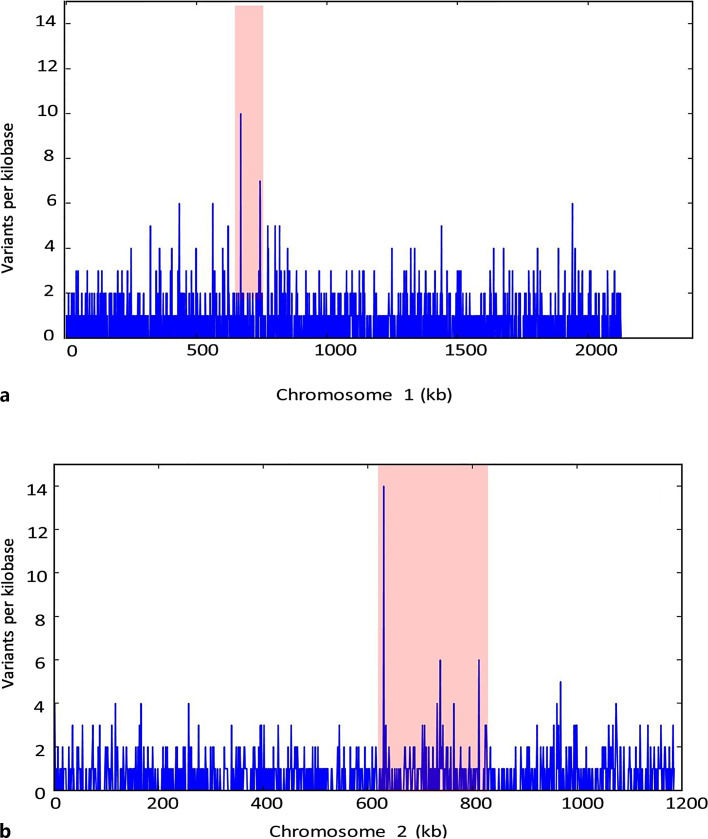
Results: The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.
Conclusions: Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.
Objective: The identification of Brucella genotypes is essential for epidemiological studies. The whole-genome sequencing is emerging as a novel tool for genetic characterization of infectious microbes. The aim of this study was to genotype Brucella melitensis isolates from Kuwait using whole-genome sequencing and variant analysis of the sequence data.
Methods: DNA was purified from 15 heat-inactivated B. melitensis isolates and used to prepare sequencing libraries employing Nextera XT DNA Sample Preparation Kit (Illumina San Diego, CA, USA) and sequenced on a MiSeq (Illumina). The sequence files were aligned to three biovars of B. melitensis, i.e., biovar 1 str. 16M, biovar 2 str. 63/9, and biovar 3 str. Ether. The alignment and variant calling were performed using "bwa-mem" and SAMtools/VCFtools, respectively.
Results: The genome size of all the isolates was around 3.3 mega base pairs and resembled B. melitensis biovar 2. Single-nucleotide polymorphisms (SNPs), insertions, and deletions (indels) were spread all over the genome; but 138 SNPs were common among the 14 isolates, supporting the same ancestral origin. A neighbor-joining tree analysis identified isolate 2 as an outlier. In addition, SNPs (2-478) specific to each isolate were also identified, which divided the B. melitensis biovar 2 into two major groups/genotypes. A further analysis showed that the Kuwaiti isolates of the present study shared phylogeny mainly with strains from the Middle Eastern countries.
Conclusions: Among the 15 studied isolates from Kuwait, biovar 2 is the most prevalent biovar of B. melitensis. Furthermore, isolate-specific genetic variations were identified, which may be useful in epidemiological investigations.
期刊介绍:
''Medical Principles and Practice'', as the journal of the Health Sciences Centre, Kuwait University, aims to be a publication of international repute that will be a medium for dissemination and exchange of scientific knowledge in the health sciences.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
