Exploring Inhibition Mechanisms in Wildtype and T315I BCR-ABL1: An In Silico Approach Integrating Virtual Screening, MD Simulations, and MM-GBSA Analysis
{"title":"Exploring Inhibition Mechanisms in Wildtype and T315I BCR-ABL1: An In Silico Approach Integrating Virtual Screening, MD Simulations, and MM-GBSA Analysis","authors":"Ozlen Balta, Ercument Yilmaz, Gizem Tatar Yilmaz","doi":"10.1002/jcc.27545","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The BCR-ABL tyrosine kinase which is responsible for the pathogenesis of chronic myeloid leukemia (CML), has emerged as a promising therapeutic target. To address this issue, we employed a comprehensive computational approach integrating virtual screening, molecular dynamics (MD) simulations, and MM-GBSA (Molecular Mechanics/Generalized Born Surface Area) analysis to identify potential inhibitors and elucidate their binding mechanisms. Initially, virtual screening was conducted on 994 compounds from the ZINC database and, these compounds were docked against wildtype and T315I mutant ABL1 for the Type I and Type II ABL1 kinase inhibition mechanisms. In our molecular docking analysis for Type I inhibition, compound 911 demonstrated notable affinity towards the wildtype ABL1, with a binding energy of −14.91 kcal/mol, while compound 972 showed significant binding affinity towards the mutant ABL1, with a binding energy of −14.27 kcal/mol. In the Type II inhibition mechanism, the compounds with the highest binding affinity were compound 261 in wildtype ABL1 with −17.05 kcal/mol binding energy and compound 966 to the mutant ABL1 with a binding energy of −16.29 kcal/mol. Furthermore, analyses of MD simulations and MM/GBSA binding free energy (ΔG) were performed for target proteins with compounds, that exhibited the most favorable binding affinities with target proteins. The selected hit compounds showed ΔG scores ranging from −118.09 to −74.85 kJ/mol in both wildtype and mutant ABL1. Considering all in silico studies performed, it can be inferred that the identified molecules hold promise as potential candidates for drug design aimed at targeting CML.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27545","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
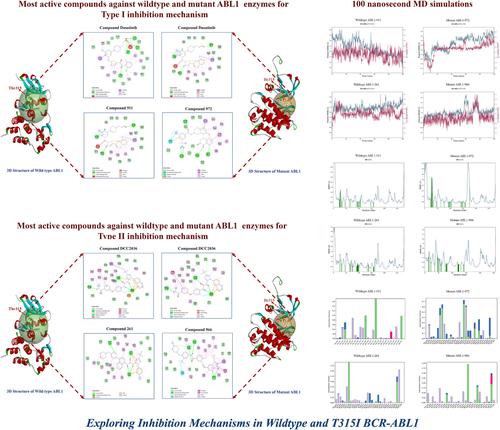
The BCR-ABL tyrosine kinase which is responsible for the pathogenesis of chronic myeloid leukemia (CML), has emerged as a promising therapeutic target. To address this issue, we employed a comprehensive computational approach integrating virtual screening, molecular dynamics (MD) simulations, and MM-GBSA (Molecular Mechanics/Generalized Born Surface Area) analysis to identify potential inhibitors and elucidate their binding mechanisms. Initially, virtual screening was conducted on 994 compounds from the ZINC database and, these compounds were docked against wildtype and T315I mutant ABL1 for the Type I and Type II ABL1 kinase inhibition mechanisms. In our molecular docking analysis for Type I inhibition, compound 911 demonstrated notable affinity towards the wildtype ABL1, with a binding energy of −14.91 kcal/mol, while compound 972 showed significant binding affinity towards the mutant ABL1, with a binding energy of −14.27 kcal/mol. In the Type II inhibition mechanism, the compounds with the highest binding affinity were compound 261 in wildtype ABL1 with −17.05 kcal/mol binding energy and compound 966 to the mutant ABL1 with a binding energy of −16.29 kcal/mol. Furthermore, analyses of MD simulations and MM/GBSA binding free energy (ΔG) were performed for target proteins with compounds, that exhibited the most favorable binding affinities with target proteins. The selected hit compounds showed ΔG scores ranging from −118.09 to −74.85 kJ/mol in both wildtype and mutant ABL1. Considering all in silico studies performed, it can be inferred that the identified molecules hold promise as potential candidates for drug design aimed at targeting CML.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
