{"title":"Modulating the oxygen affinity of porphyrins with metals, ligands, and functional groups: A DFT study","authors":"Sebastian Ovalle, Cecile Malardier-Jugroot","doi":"10.1002/jcc.27505","DOIUrl":null,"url":null,"abstract":"<p>The interaction between different metals (M), axial ligands (L), and ring substituents (R) in porphyrins was investigated using density functional theory. Different combinations of iron and cobalt as metal centers; imidazole, chlorine, and an n-heterocyclic carbene (NHC) as axial ligands, and unsubstituted, octaethyl-, and tetraphenyl-porphyrins were explored in their low, intermediate, and high-spin states, alongside oxygen affinity. Remarkably, the n-heterocyclic carbene enhanced the affinity of cobalt porphyrins to oxygen, with binding energies on average 4.4 kcal mol<sup>−1</sup> higher than FeP with the same ligand, and 0.78 kcal mol<sup>−1</sup> higher than FeP with imidazole. The planarity of the iron tetraphenyl porphyrin with imidazole compared to its ruffled cobalt counterpart is noteworthy in both oxy- and deoxy-forms, highlighting imidazole's stabilizing influence on the porphyrin structure, particularly iron porphyrins, alongside imidazole's stabilizing effect on the affinity to O<sub>2</sub>. Despite the significant non-planarity induced by NHC as an axial ligand -regardless of the metal or ring substituent used-, it did not hinder the affinity of CoP to O<sub>2</sub> (14.26 kcal mol<sup>−1</sup>, on average) as it did with the FeP with NHC (9.88 kcal mol<sup>−1</sup>, on average). Cobalt porphyrins with n-heterocyclic carbene ligands show promising potential for O<sub>2</sub> activation or oxygen transport applications. The results show the complex interactions between the different parts of metalloporphyrins and highlight the capability of tailoring their affinity to O<sub>2</sub>. It also exemplifies the stabilizing effect of imidazole on the porphyrins, providing a very narrow range of binding energies and smaller differences in their geometries.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-12-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27505","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27505","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
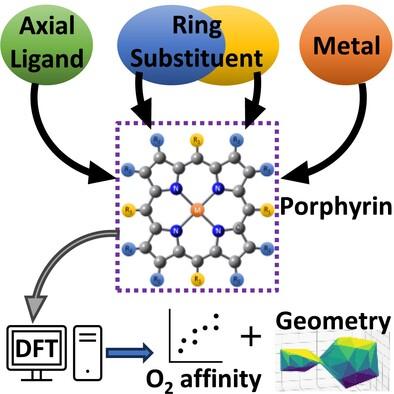
Abstract
The interaction between different metals (M), axial ligands (L), and ring substituents (R) in porphyrins was investigated using density functional theory. Different combinations of iron and cobalt as metal centers; imidazole, chlorine, and an n-heterocyclic carbene (NHC) as axial ligands, and unsubstituted, octaethyl-, and tetraphenyl-porphyrins were explored in their low, intermediate, and high-spin states, alongside oxygen affinity. Remarkably, the n-heterocyclic carbene enhanced the affinity of cobalt porphyrins to oxygen, with binding energies on average 4.4 kcal mol−1 higher than FeP with the same ligand, and 0.78 kcal mol−1 higher than FeP with imidazole. The planarity of the iron tetraphenyl porphyrin with imidazole compared to its ruffled cobalt counterpart is noteworthy in both oxy- and deoxy-forms, highlighting imidazole's stabilizing influence on the porphyrin structure, particularly iron porphyrins, alongside imidazole's stabilizing effect on the affinity to O2. Despite the significant non-planarity induced by NHC as an axial ligand -regardless of the metal or ring substituent used-, it did not hinder the affinity of CoP to O2 (14.26 kcal mol−1, on average) as it did with the FeP with NHC (9.88 kcal mol−1, on average). Cobalt porphyrins with n-heterocyclic carbene ligands show promising potential for O2 activation or oxygen transport applications. The results show the complex interactions between the different parts of metalloporphyrins and highlight the capability of tailoring their affinity to O2. It also exemplifies the stabilizing effect of imidazole on the porphyrins, providing a very narrow range of binding energies and smaller differences in their geometries.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
