Oliver J. Conquest, Yijiao Jiang, Catherine Stampfl
{"title":"CoTCNQ as a Catalyst for CO2 Electroreduction: A First Principles r2SCAN Meta-GGA Investigation","authors":"Oliver J. Conquest, Yijiao Jiang, Catherine Stampfl","doi":"10.1002/jcc.27528","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Using first principles calculations we investigate cobalt-coordinated tetracyanoquinodimethane (R-CoTCNQ) as a potential catalyst for the CO<sub>2</sub> electroreduction reaction (CO<sub>2</sub>ERR). We determine that exchange–correlation functionals beyond the generalized gradient approximation (GGA) are required to accurately describe the spin properties of R-CoTCNQ, therefore, the meta-GGA r<sup>2</sup>SCAN functional is used in this study. The free energy CO<sub>2</sub>ERR reaction pathways are calculated for the reduced catalyst ([R-CoTCNQ]<sup>−1<i>e</i></sup>) with reaction products HCOOH and HCHO predicted depending on our choice of electrode potential. Calculations are also performed for [R-CoTCNQ]<sup>−1<i>e</i></sup> supported on a H-terminated diamond (1 1 0) surface with reaction pathways being qualitatively similar to the [R-CoTCNQ]<sup>−1<i>e</i></sup> monolayer. The inclusion of boron-doping in the diamond support shows a slightly improved CO<sub>2</sub>ERR reaction pathway. Furthermore, structurally, supported R-CoTCNQ provide a high specific area of active Co active sites and could be promising catalysts for future experimental consideration.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27528","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
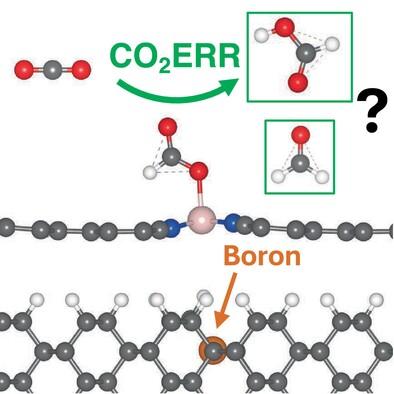
Using first principles calculations we investigate cobalt-coordinated tetracyanoquinodimethane (R-CoTCNQ) as a potential catalyst for the CO2 electroreduction reaction (CO2ERR). We determine that exchange–correlation functionals beyond the generalized gradient approximation (GGA) are required to accurately describe the spin properties of R-CoTCNQ, therefore, the meta-GGA r2SCAN functional is used in this study. The free energy CO2ERR reaction pathways are calculated for the reduced catalyst ([R-CoTCNQ]−1e) with reaction products HCOOH and HCHO predicted depending on our choice of electrode potential. Calculations are also performed for [R-CoTCNQ]−1e supported on a H-terminated diamond (1 1 0) surface with reaction pathways being qualitatively similar to the [R-CoTCNQ]−1e monolayer. The inclusion of boron-doping in the diamond support shows a slightly improved CO2ERR reaction pathway. Furthermore, structurally, supported R-CoTCNQ provide a high specific area of active Co active sites and could be promising catalysts for future experimental consideration.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
