{"title":"Groupy: An Open-Source Toolkit for Molecular Simulation and Property Calculation","authors":"Ruichen Liu, Li Wang, Xiangwen Zhang, Guozhu Li","doi":"10.1002/jcc.27527","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>In this work, an open-source, versatile, and flexible code named Groupy is present for calculating various molecular properties and preparing input files of molecular simulation software such as Gaussian. This code requires only SMILES as input, but can output many new useful data and files in multiple formats. The output information is clear and easy to read. The tips to the users are very detailed and easy to follow when using. Message passing interface (MPI) parallelization is supported to reduce computing time when the properties of a large number of molecules are calculated. Groupy not only supports the calculation of molecular properties using the traditional group contribution method, but also directly outputs the group-contribution-style molecular fingerprints for machine learning. The code has strong extensibility, which can be used as an external library to build other programs. We hope that Groupy brings great convenience to both computational and experimental chemists in their daily research. The code of Groupy can be freely obtained at https://github.com/47-5/Groupy</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-12-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27527","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
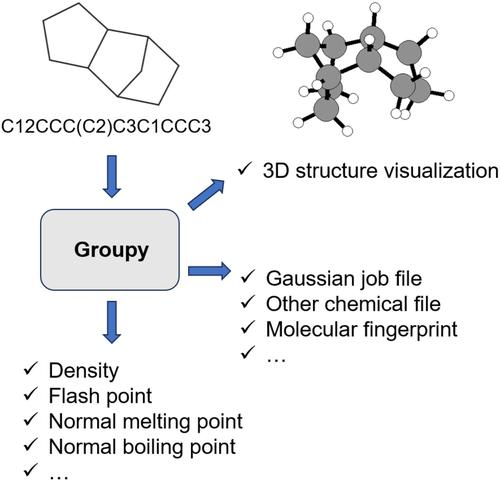
In this work, an open-source, versatile, and flexible code named Groupy is present for calculating various molecular properties and preparing input files of molecular simulation software such as Gaussian. This code requires only SMILES as input, but can output many new useful data and files in multiple formats. The output information is clear and easy to read. The tips to the users are very detailed and easy to follow when using. Message passing interface (MPI) parallelization is supported to reduce computing time when the properties of a large number of molecules are calculated. Groupy not only supports the calculation of molecular properties using the traditional group contribution method, but also directly outputs the group-contribution-style molecular fingerprints for machine learning. The code has strong extensibility, which can be used as an external library to build other programs. We hope that Groupy brings great convenience to both computational and experimental chemists in their daily research. The code of Groupy can be freely obtained at https://github.com/47-5/Groupy
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
