Shashank V. Rao, Dimitrios Maganas, Kantharuban Sivalingam, Mihail Atanasov, Frank Neese
{"title":"Extended Active Space Ab Initio Ligand Field Theory: Applications to Transition-Metal Ions","authors":"Shashank V. Rao, Dimitrios Maganas, Kantharuban Sivalingam, Mihail Atanasov, Frank Neese","doi":"10.1021/acs.inorgchem.4c03893","DOIUrl":null,"url":null,"abstract":"Ligand field theory (LFT) is one of the cornerstones of coordination chemistry since it provides a conceptual framework in which a great many properties of d- and f-element compounds can be discussed. While LFT serves as a powerful qualitative guide, it is not a tool for quantitative predictions on individual compounds since it incorporates semiempirical parameters that must be fitted to experiment. One way to connect the realms of first-principles electronic structure theory that has emerged as particularly powerful over the past decade is the ab initio ligand field theory (AILFT). The original formulation of this method involved the extraction of LFT parameters by fitting the ligand field Hamiltonian to a complete active space self-consistent field (CASSCF) Hamiltonian. The extraction was shown to be unique provided that the active space consists of 5/7 metal d/f-based molecular orbitals (MOs). Subsequent improvements have involved incorporating dynamical correlation using second-order N-electron valence state perturbation theory (NEVPT2) or second-order dynamical correlation dressed complete active space (DCDCAS). However, the limitation of past approaches is that the method requires a minimal space of 5/7 metal d- or f-based molecular orbitals. This leads to a number of limitations: (1) neglect of radial or semicore correlation would arise from the effect of a second d-shell or an sp-shell in the active space, (2) a more balanced description of metal–ligand bond covalency is lacking because the bonding ligand-based counterparts of the metal d/f orbitals are not in the active space. This usually leads to an exaggerated ionicity of the M–L bonds. In this work, we present an extended active space AILFT (esAILFT) that circumvents these limitations and is, in principle, applicable to arbitrary active spaces, as long as these contain the 5/7 metal d/f-based MOs as a subset. esAILFT was implemented in a development version of the ORCA software package. In order to help with the application of the new method, various criteria for active space extension were explored for 3d, 4d, and 5d transition-metal ions with varying charge. An interpretation of the trends in the Racah B parameter for these ions is also presented as a demonstration of the capabilities of esAILFT.","PeriodicalId":40,"journal":{"name":"Inorganic Chemistry","volume":"54 1","pages":""},"PeriodicalIF":4.7000,"publicationDate":"2024-12-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.inorgchem.4c03893","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
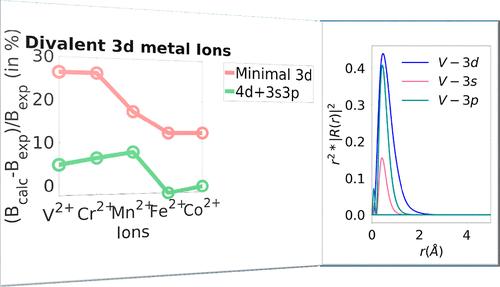
Ligand field theory (LFT) is one of the cornerstones of coordination chemistry since it provides a conceptual framework in which a great many properties of d- and f-element compounds can be discussed. While LFT serves as a powerful qualitative guide, it is not a tool for quantitative predictions on individual compounds since it incorporates semiempirical parameters that must be fitted to experiment. One way to connect the realms of first-principles electronic structure theory that has emerged as particularly powerful over the past decade is the ab initio ligand field theory (AILFT). The original formulation of this method involved the extraction of LFT parameters by fitting the ligand field Hamiltonian to a complete active space self-consistent field (CASSCF) Hamiltonian. The extraction was shown to be unique provided that the active space consists of 5/7 metal d/f-based molecular orbitals (MOs). Subsequent improvements have involved incorporating dynamical correlation using second-order N-electron valence state perturbation theory (NEVPT2) or second-order dynamical correlation dressed complete active space (DCDCAS). However, the limitation of past approaches is that the method requires a minimal space of 5/7 metal d- or f-based molecular orbitals. This leads to a number of limitations: (1) neglect of radial or semicore correlation would arise from the effect of a second d-shell or an sp-shell in the active space, (2) a more balanced description of metal–ligand bond covalency is lacking because the bonding ligand-based counterparts of the metal d/f orbitals are not in the active space. This usually leads to an exaggerated ionicity of the M–L bonds. In this work, we present an extended active space AILFT (esAILFT) that circumvents these limitations and is, in principle, applicable to arbitrary active spaces, as long as these contain the 5/7 metal d/f-based MOs as a subset. esAILFT was implemented in a development version of the ORCA software package. In order to help with the application of the new method, various criteria for active space extension were explored for 3d, 4d, and 5d transition-metal ions with varying charge. An interpretation of the trends in the Racah B parameter for these ions is also presented as a demonstration of the capabilities of esAILFT.
期刊介绍:
Inorganic Chemistry publishes fundamental studies in all phases of inorganic chemistry. Coverage includes experimental and theoretical reports on quantitative studies of structure and thermodynamics, kinetics, mechanisms of inorganic reactions, bioinorganic chemistry, and relevant aspects of organometallic chemistry, solid-state phenomena, and chemical bonding theory. Emphasis is placed on the synthesis, structure, thermodynamics, reactivity, spectroscopy, and bonding properties of significant new and known compounds.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
