{"title":"Comprehensive Analysis of Deuterium Isotope Effects on Ionic H3O+…π Interactions Using Multi-Component Quantum Mechanics Methods","authors":"Taro Udagawa, Yusuke Kanematsu, Takayoshi Ishimoto, Masanori Tachikawa","doi":"10.1002/jcc.70000","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Deuterium isotope effects on interaction energies and geometrical parameters in several H<sub>3</sub>O<sup>+</sup>(D<sub>3</sub>O<sup>+</sup>)<sup>…</sup>ene and H<sub>3</sub>O+(D<sub>3</sub>O<sup>+</sup>)<sup>…</sup>yne complexes, which involve O-H(D)<sup>…</sup>π interactions, have been analyzed using the MP2 level of the multi-component molecular orbital method (MC_MP2), which can incorporate the nuclear quantum effects of light nuclei, such as protons and deuterons. The MC_MP2 calculations revealed that D<sub>3</sub>O<sup>+</sup> replacement reduced the interaction energies of the complexes and induced changes in geometrical parameters. In addition, natural energy decomposition analysis (NEDA) revealed a strong correlation between the H/D isotope effects on the H/D<sup>…</sup>π distances and on each energy component.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-12-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70000","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
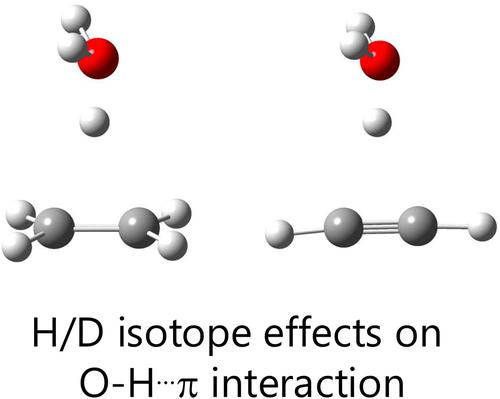
Abstract
Deuterium isotope effects on interaction energies and geometrical parameters in several H3O+(D3O+)…ene and H3O+(D3O+)…yne complexes, which involve O-H(D)…π interactions, have been analyzed using the MP2 level of the multi-component molecular orbital method (MC_MP2), which can incorporate the nuclear quantum effects of light nuclei, such as protons and deuterons. The MC_MP2 calculations revealed that D3O+ replacement reduced the interaction energies of the complexes and induced changes in geometrical parameters. In addition, natural energy decomposition analysis (NEDA) revealed a strong correlation between the H/D isotope effects on the H/D…π distances and on each energy component.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
