Adeeba Afrah, Michael A Finkel, Carolina Fonseca, Marianne Tomiyoshi Asato, M Susan Jay, Athina Pappas, Shashikala B Gowda, Allison Jay
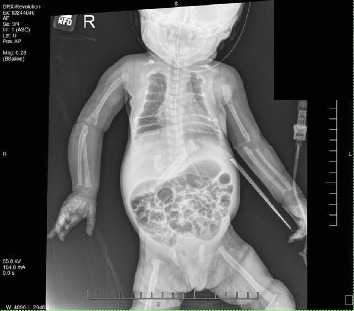
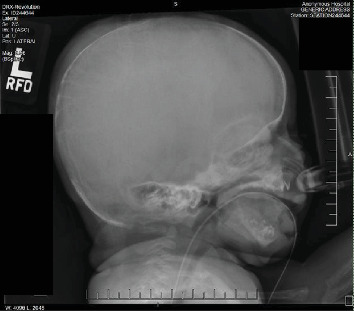
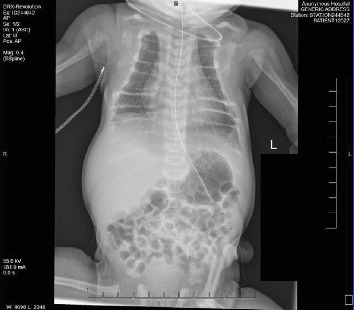
{"title":"Demineralization of Osseous Structures as Presentation of a Rare Genetic Disorder That Is Associated With a High Rate of Mortality.","authors":"Adeeba Afrah, Michael A Finkel, Carolina Fonseca, Marianne Tomiyoshi Asato, M Susan Jay, Athina Pappas, Shashikala B Gowda, Allison Jay","doi":"10.1155/crie/6063059","DOIUrl":null,"url":null,"abstract":"<p><p><b>Objectives:</b> Describe the details of the clinical presentation, diagnostic challenges, and management of a female neonate with neonatal severe hyperparathyroidism (NSHPT). <b>Methods:</b> This case report was developed from a retrospective chart review. The female infant was born to consanguineous parents-first cousins, with multiple prenatal concerns, including gestational diabetes, intrauterine growth restriction, polyhydramnios, and suspicion of a hypoplastic left atrium on prenatal echocardiogram (ECHO). Following a planned C-section at 37 weeks gestation, the neonate exhibited moderate respiratory distress with subcostal retractions. On physical examination, craniotabes, a bell-shaped chest, and a continuous machinery-type murmur were noted. <b>Results:</b> Evaluation at birth revealed a large Patent Ductus Arteriosus and significant demineralization of skeletal structures with atypical rib morphology. Lab work at 24 h of life (HOL) showed elevated serum calcium level (14.3 mg/dL), ionized calcium-iCal (2.32 mmol/L), and normal 25-OH Vitamin D (54.2 ng/mL). A comprehensive skeletal survey uncovered generalized osteopenia, metaphyseal lucencies, and evidence of healing fractures. Repeat lab work at 43 HOL, showed serum calcium of 18.0 mg/dL, iCal 2.67 mmol/L, and elevated parathyroid hormone (PTH) of 2116 pg/mL. Diagnosis of NSHPT was established based on laboratory findings. Molecular testing confirmed a homozygous variant (c.1744T >A; p.Cys582Ser) in the calcium-sensing receptor (CaSR) gene which confirmed the diagnosis of NSHPT. NSHPT, a rare genetic disorder associated with high mortality rates, is often caused by inactivating CaSR gene variants. The patient's family history revealed a strong correlation with familial hypocalciuric hypercalcemia (FHH), a benign condition associated with asymptomatic hypercalcemia, normal to minimally elevated parathyroid level, and hypocalciuria, it is caused by heterozygous inactivating mutations in the CaSR gene. Treatment of NSHPT typically involves total or subtotal parathyroidectomy; however, initial medical intervention is often necessary. In this case, the neonate underwent medical treatment with calcitonin, furosemide to help facilitate renal clearance of calcium, and intravenous fluids before a successful parathyroidectomy. <b>Conclusions:</b> This case accentuates the importance of considering rare genetic disorders in neonates with complex clinical presentations and affirms the need for comprehensive counseling and education, particularly in consanguineous parents, to address familial implications and guide appropriate interventions.</p>","PeriodicalId":9621,"journal":{"name":"Case Reports in Endocrinology","volume":"2024 ","pages":"6063059"},"PeriodicalIF":0.9000,"publicationDate":"2024-12-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11658845/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/crie/6063059","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Objectives: Describe the details of the clinical presentation, diagnostic challenges, and management of a female neonate with neonatal severe hyperparathyroidism (NSHPT). Methods: This case report was developed from a retrospective chart review. The female infant was born to consanguineous parents-first cousins, with multiple prenatal concerns, including gestational diabetes, intrauterine growth restriction, polyhydramnios, and suspicion of a hypoplastic left atrium on prenatal echocardiogram (ECHO). Following a planned C-section at 37 weeks gestation, the neonate exhibited moderate respiratory distress with subcostal retractions. On physical examination, craniotabes, a bell-shaped chest, and a continuous machinery-type murmur were noted. Results: Evaluation at birth revealed a large Patent Ductus Arteriosus and significant demineralization of skeletal structures with atypical rib morphology. Lab work at 24 h of life (HOL) showed elevated serum calcium level (14.3 mg/dL), ionized calcium-iCal (2.32 mmol/L), and normal 25-OH Vitamin D (54.2 ng/mL). A comprehensive skeletal survey uncovered generalized osteopenia, metaphyseal lucencies, and evidence of healing fractures. Repeat lab work at 43 HOL, showed serum calcium of 18.0 mg/dL, iCal 2.67 mmol/L, and elevated parathyroid hormone (PTH) of 2116 pg/mL. Diagnosis of NSHPT was established based on laboratory findings. Molecular testing confirmed a homozygous variant (c.1744T >A; p.Cys582Ser) in the calcium-sensing receptor (CaSR) gene which confirmed the diagnosis of NSHPT. NSHPT, a rare genetic disorder associated with high mortality rates, is often caused by inactivating CaSR gene variants. The patient's family history revealed a strong correlation with familial hypocalciuric hypercalcemia (FHH), a benign condition associated with asymptomatic hypercalcemia, normal to minimally elevated parathyroid level, and hypocalciuria, it is caused by heterozygous inactivating mutations in the CaSR gene. Treatment of NSHPT typically involves total or subtotal parathyroidectomy; however, initial medical intervention is often necessary. In this case, the neonate underwent medical treatment with calcitonin, furosemide to help facilitate renal clearance of calcium, and intravenous fluids before a successful parathyroidectomy. Conclusions: This case accentuates the importance of considering rare genetic disorders in neonates with complex clinical presentations and affirms the need for comprehensive counseling and education, particularly in consanguineous parents, to address familial implications and guide appropriate interventions.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
