Sourabh Kumar, Gokay Adabasi, Mehmet Z. Baykara, Ashlie Martini
{"title":"Modulating the Electronic Properties of Orthorhombic Mo2C Surfaces with Strain and Defects: Insights from First-Principles Calculations","authors":"Sourabh Kumar, Gokay Adabasi, Mehmet Z. Baykara, Ashlie Martini","doi":"10.1021/acs.jpcc.4c07211","DOIUrl":null,"url":null,"abstract":"Mo<sub>2</sub>C is an efficient and cost-effective catalyst for hydrogenation reactions that are crucial for chemical synthesis and renewable energy applications. In this study, we investigate the electronic and adsorption properties of orthorhombic Mo<sub>2</sub>C (001) with three different surface terminations using density functional theory. By introducing Mo and C vacancies and substituting Mo with Ti, we evaluate the effect of defects on the electron localization function (ELF), projected density of states, and hydrogen adsorption behavior. The results show that Mo atom vacancies significantly disrupt the ELF distribution, while C atom vacancies and Ti substitutions have little effect. Tensile or compressive strain applied to the surfaces modulates the ELF for surfaces with Mo defects but has little effect on systems with C vacancies or Ti substitutions. We also examine how defects and strain affect hydrogen adsorption on the Mo<sub>2</sub>C surfaces to understand the potential effect on catalytic performance. The findings of this study highlight the importance of defect and strain conditions in the catalytic efficiency of orthorhombic Mo<sub>2</sub>C and offer valuable insight into designing strain- and defect-engineered catalysts with enhanced hydrogen adsorption and desorption properties, paving the way for more efficient and selective hydrogenation processes.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"20 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-12-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c07211","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
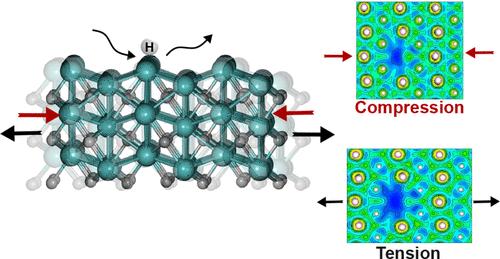
Mo2C is an efficient and cost-effective catalyst for hydrogenation reactions that are crucial for chemical synthesis and renewable energy applications. In this study, we investigate the electronic and adsorption properties of orthorhombic Mo2C (001) with three different surface terminations using density functional theory. By introducing Mo and C vacancies and substituting Mo with Ti, we evaluate the effect of defects on the electron localization function (ELF), projected density of states, and hydrogen adsorption behavior. The results show that Mo atom vacancies significantly disrupt the ELF distribution, while C atom vacancies and Ti substitutions have little effect. Tensile or compressive strain applied to the surfaces modulates the ELF for surfaces with Mo defects but has little effect on systems with C vacancies or Ti substitutions. We also examine how defects and strain affect hydrogen adsorption on the Mo2C surfaces to understand the potential effect on catalytic performance. The findings of this study highlight the importance of defect and strain conditions in the catalytic efficiency of orthorhombic Mo2C and offer valuable insight into designing strain- and defect-engineered catalysts with enhanced hydrogen adsorption and desorption properties, paving the way for more efficient and selective hydrogenation processes.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
