Alexandra Chera, Mircea Stancu-Cretu, Nicolae Radu Zabet, Octavian Bucur
{"title":"Shedding light on DNA methylation and its clinical implications: the impact of long-read-based nanopore technology.","authors":"Alexandra Chera, Mircea Stancu-Cretu, Nicolae Radu Zabet, Octavian Bucur","doi":"10.1186/s13072-024-00558-2","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation is an essential epigenetic mechanism for regulation of gene expression, through which many physiological (X-chromosome inactivation, genetic imprinting, chromatin structure and miRNA regulation, genome defense, silencing of transposable elements) and pathological processes (cancer and repetitive sequences-associated diseases) are regulated. Nanopore sequencing has emerged as a novel technique that can analyze long strands of DNA (long-read sequencing) without chemically treating the DNA. Interestingly, nanopore sequencing can also extract epigenetic status of the nucleotides (including both 5-Methylcytosine and 5-hydroxyMethylcytosine), and a large variety of bioinformatic tools have been developed for improving its detection properties. Out of all genomic regions, long read sequencing provides advantages in studying repetitive elements, which are difficult to characterize through other sequencing methods. Transposable elements are repetitive regions of the genome that are silenced and usually display high levels of DNA methylation. Their demethylation and activation have been observed in many cancers. Due to their repetitive nature, it is challenging to accurately estimate DNA methylation levels within transposable elements using short sequencing technologies. The advantage to sequence native DNA (without PCR amplification biases or harsh bisulfite treatment) and long and ultra long reads coupled with epigenetic states of the DNA allows to accurately estimate DNA methylation levels in transposable elements. This is a big step forward for epigenomic studies, and unsolved questions regarding gene expression and transposable elements silencing through DNA methylation can now be answered.</p>","PeriodicalId":49253,"journal":{"name":"Epigenetics & Chromatin","volume":"17 1","pages":"39"},"PeriodicalIF":3.5000,"publicationDate":"2024-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11684317/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epigenetics & Chromatin","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13072-024-00558-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
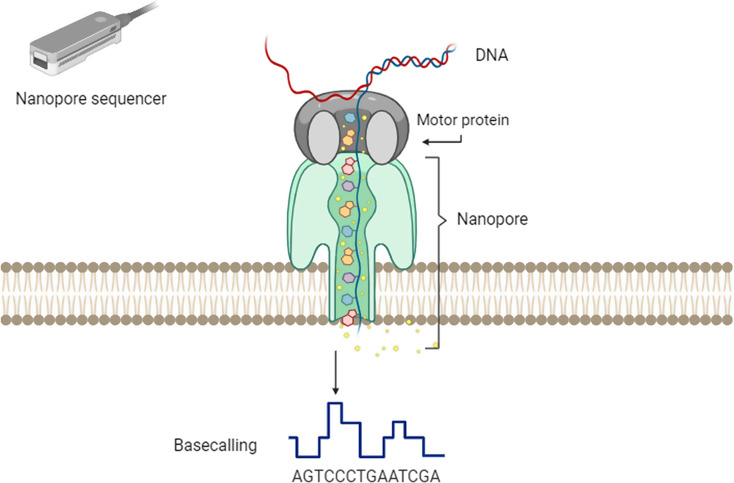
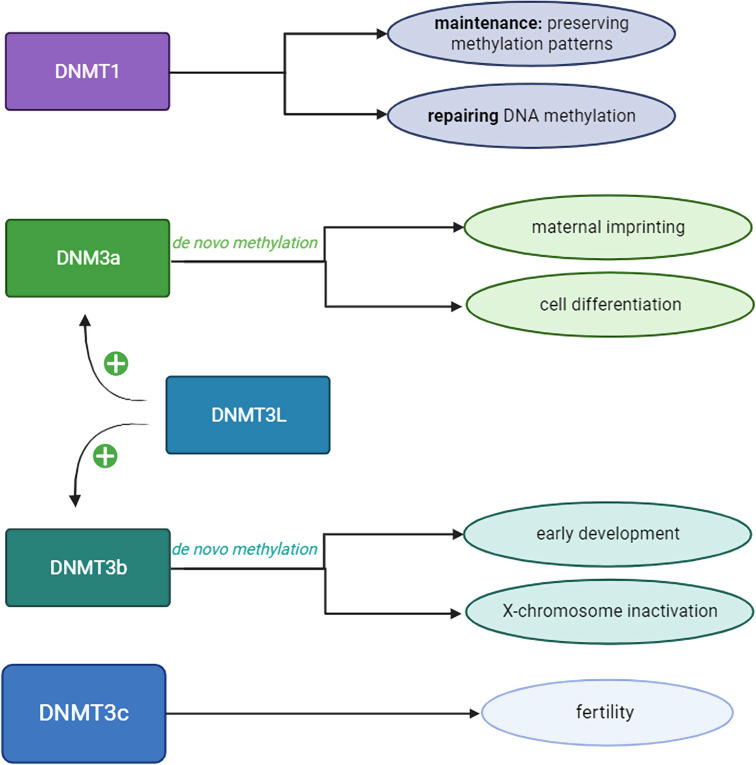
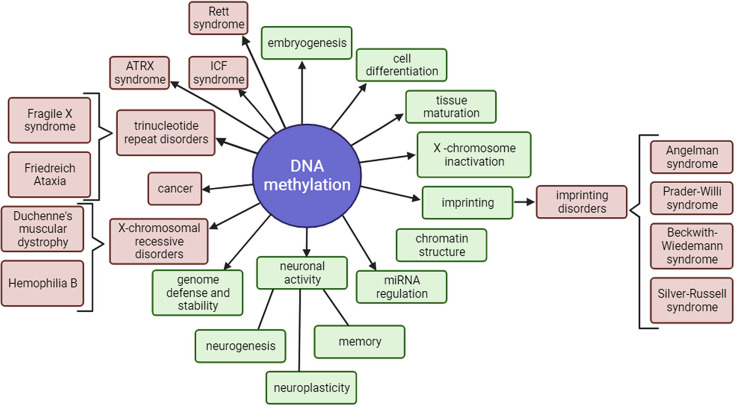
DNA methylation is an essential epigenetic mechanism for regulation of gene expression, through which many physiological (X-chromosome inactivation, genetic imprinting, chromatin structure and miRNA regulation, genome defense, silencing of transposable elements) and pathological processes (cancer and repetitive sequences-associated diseases) are regulated. Nanopore sequencing has emerged as a novel technique that can analyze long strands of DNA (long-read sequencing) without chemically treating the DNA. Interestingly, nanopore sequencing can also extract epigenetic status of the nucleotides (including both 5-Methylcytosine and 5-hydroxyMethylcytosine), and a large variety of bioinformatic tools have been developed for improving its detection properties. Out of all genomic regions, long read sequencing provides advantages in studying repetitive elements, which are difficult to characterize through other sequencing methods. Transposable elements are repetitive regions of the genome that are silenced and usually display high levels of DNA methylation. Their demethylation and activation have been observed in many cancers. Due to their repetitive nature, it is challenging to accurately estimate DNA methylation levels within transposable elements using short sequencing technologies. The advantage to sequence native DNA (without PCR amplification biases or harsh bisulfite treatment) and long and ultra long reads coupled with epigenetic states of the DNA allows to accurately estimate DNA methylation levels in transposable elements. This is a big step forward for epigenomic studies, and unsolved questions regarding gene expression and transposable elements silencing through DNA methylation can now be answered.
DNA 甲基化是调控基因表达的重要表观遗传机制,许多生理过程(X 染色体失活、基因印记、染色质结构和 miRNA 调控、基因组防御、转座元件沉默)和病理过程(癌症和重复序列相关疾病)都是通过这种机制调控的。纳米孔测序是一种新型技术,无需对 DNA 进行化学处理即可分析长链 DNA(长读取测序)。有趣的是,纳米孔测序技术还能提取核苷酸(包括 5-甲基胞嘧啶和 5-羟基甲基胞嘧啶)的表观遗传学状态,目前已开发出大量生物信息学工具来改进其检测性能。在所有基因组区域中,长读测序在研究其他测序方法难以表征的重复性元件方面具有优势。可转座元件是基因组中被沉默的重复区域,通常表现出高水平的 DNA 甲基化。在许多癌症中都观察到了它们的去甲基化和激活。由于其重复性,使用短测序技术准确估计可转座元件内的 DNA 甲基化水平具有挑战性。对原生 DNA 进行测序(没有 PCR 扩增偏差或苛刻的亚硫酸氢盐处理)以及长读数和超长读数与 DNA 的表观遗传状态相结合的优势,可以准确估计转座元件中的 DNA 甲基化水平。这是表观基因组研究向前迈出的一大步,有关基因表达和转座元件通过 DNA 甲基化沉默的未决问题现在可以得到解答了。
期刊介绍:
Epigenetics & Chromatin is a peer-reviewed, open access, online journal that publishes research, and reviews, providing novel insights into epigenetic inheritance and chromatin-based interactions. The journal aims to understand how gene and chromosomal elements are regulated and their activities maintained during processes such as cell division, differentiation and environmental alteration.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
