Vaibhav Khanna, Bikash Kanungo, Vikram Gavini, Ambuj Tewari, Paul M. Zimmerman
{"title":"Examining the Impact of Local Constraint Violations on Energy Computations in DFT","authors":"Vaibhav Khanna, Bikash Kanungo, Vikram Gavini, Ambuj Tewari, Paul M. Zimmerman","doi":"10.1002/jcc.70005","DOIUrl":null,"url":null,"abstract":"<p>This work examines the impact of locally imposed constraints in Density Functional Theory (DFT). Using a metric referred to as the extent of violation index (EVI), we quantify how well exchange-correlation functionals adhere to local constraints. Applying EVIs to a diverse set of molecules for GGA functionals reveals constraint violations, particularly for semi-empirical functionals. We leverage EVIs to explore potential connections between these violations and errors in chemical properties. While no correlation is observed for atomization energies, a significant statistical correlation emerges between EVIs and total energies. Similarly, the analysis of reaction energies suggests weak positive correlations for specific constraints. However, definitive conclusions about error cancellation mechanisms cannot be made at this time. These observations revealed by EVIs may be useful for consideration when designing future generations of semilocal functionals.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70005","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70005","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
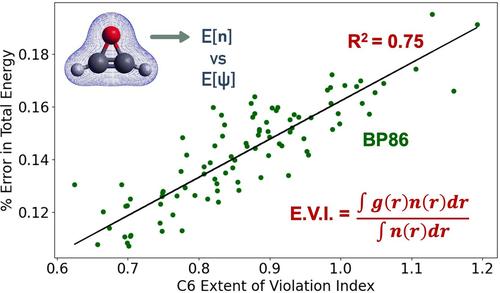
This work examines the impact of locally imposed constraints in Density Functional Theory (DFT). Using a metric referred to as the extent of violation index (EVI), we quantify how well exchange-correlation functionals adhere to local constraints. Applying EVIs to a diverse set of molecules for GGA functionals reveals constraint violations, particularly for semi-empirical functionals. We leverage EVIs to explore potential connections between these violations and errors in chemical properties. While no correlation is observed for atomization energies, a significant statistical correlation emerges between EVIs and total energies. Similarly, the analysis of reaction energies suggests weak positive correlations for specific constraints. However, definitive conclusions about error cancellation mechanisms cannot be made at this time. These observations revealed by EVIs may be useful for consideration when designing future generations of semilocal functionals.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
