{"title":"Thermodynamic Stability in Transition Metal-Hydrogen Dications: Potential Energy Curves, Spectroscopic Parameters, and Bonding for VH2+","authors":"João Gabriel Farias Romeu, Fernando R. Ornellas","doi":"10.1002/jcc.27530","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Seventeen electronic states of the dication VH<sup>2+</sup> were characterized by the SA-CASSCF/icMRCI methodology using very extended basis sets; 11 were described for the first time. Potential energy curves were constructed and the associated spectroscopic parameters evaluated. Triplet and quintet states correlating with the V<sup>2+</sup> + H channel are thermodynamic stable. For states dissociating into the channel V<sup>+</sup> + H<sup>+</sup>, avoided crossings at large distances give rise to thermodynamic metastability but do not affect the characterization of the bound region. Configuration state functions with the 3σ orbital /doubly occupied give rise to covalent contributions to the bonding; the major contribution, however, comes from the electrostatic charge-induced dipole interaction. This explains the shape and proximity of the potential energy curves beyond their equilibrium distances. Dipole moment functions and vibrationally averaged dipole moments quantify the polarity of the molecule. Spin–orbit couplings give rise to complex and dense regions of very close-lying Ω states.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27530","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
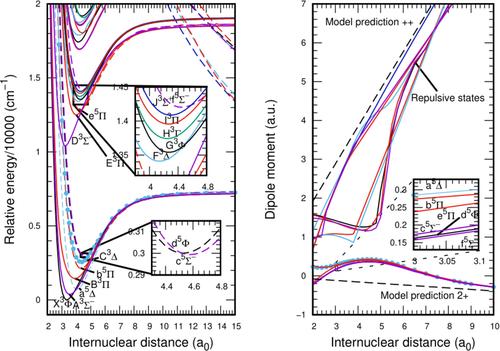
Seventeen electronic states of the dication VH2+ were characterized by the SA-CASSCF/icMRCI methodology using very extended basis sets; 11 were described for the first time. Potential energy curves were constructed and the associated spectroscopic parameters evaluated. Triplet and quintet states correlating with the V2+ + H channel are thermodynamic stable. For states dissociating into the channel V+ + H+, avoided crossings at large distances give rise to thermodynamic metastability but do not affect the characterization of the bound region. Configuration state functions with the 3σ orbital /doubly occupied give rise to covalent contributions to the bonding; the major contribution, however, comes from the electrostatic charge-induced dipole interaction. This explains the shape and proximity of the potential energy curves beyond their equilibrium distances. Dipole moment functions and vibrationally averaged dipole moments quantify the polarity of the molecule. Spin–orbit couplings give rise to complex and dense regions of very close-lying Ω states.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
