Quan Manh Phung, Takeshi Yanai, Dieter Plessers, Bert F. Sels, Robert A. Schoonheydt, Kristine Pierloot
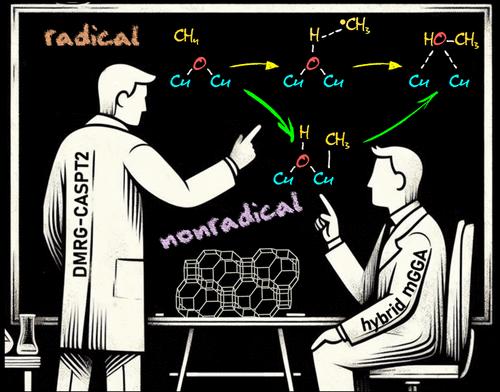
{"title":"Homolytic versus Heterolytic Methane Hydroxylation in Copper Zeolites","authors":"Quan Manh Phung, Takeshi Yanai, Dieter Plessers, Bert F. Sels, Robert A. Schoonheydt, Kristine Pierloot","doi":"10.1021/acscatal.4c06246","DOIUrl":null,"url":null,"abstract":"Oxygen-activated copper zeolites are capable of selectively converting methane to methanol at mild conditions, using a mono-oxygen bridged Cu(II) site [CuOCu]<sup>2+</sup> as the active core. Based on previous DFT reports on the [CuOCu]<sup>2+</sup> + CH<sub>4</sub> reaction a general consensus was reached concerning the methane oxidation mechanism, where the rate-limiting step involves homolytic C–H bond cleavage to form [Cu(OH)Cu]<sup>2+</sup> with a physisorbed •CH<sub>3</sub>. An alternative possibility, i.e. heterolytic H-abstraction passing through a four-center transition state to give an intermediate with a Cu–CH<sub>3</sub> bond, was given consideration only in a few recent DFT studies, but was found less favorable than radical C–H activation. In this contribution methane-to-methanol conversion by Cu–CHA is investigated using large cluster models and employing either DFT, with an extensive list of 97 functionals, or the high-level correlated DMRG/cu(4)-CASPT2 method. In all cases homolytic C–H dissociation most favorably proceeds via a (<i>S</i> = 1) transition state TS1r, whereas the transition state of heterolytic H-abstraction, TS1n, has an (<i>S</i> = 0) ground state. The DMRG/cu(4)-CASPT2 results convincingly point to the heterolytic route, with a calculated activation enthalpy of 12.3 kcal/mol, as compared to 21.1 kcal/mol for homolytic C–H dissociation. In contrast, the results obtained with DFT are strongly functional dependent. Conform with previous DFT studies, homolytic H-abstraction is preferred by the B3LYP functional (almost exclusively used in previous cluster model studies). However, many other functionals, hybrid meta-GGA functionals in particular, agree with DMRG/cu(4)-CASPT2 on heterolytic C–H activation. The present results reopen the debate on the general validity of the radical rebound mechanism for methane hydroxylation by a [CuOCu]<sup>2+</sup> core in copper zeolites and also highlight the need for caution when relying on a specific DFT functional to elucidate oxidation reaction mechanisms in metal-based catalytic systems.","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"28 1","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2025-01-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acscatal.4c06246","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Oxygen-activated copper zeolites are capable of selectively converting methane to methanol at mild conditions, using a mono-oxygen bridged Cu(II) site [CuOCu]2+ as the active core. Based on previous DFT reports on the [CuOCu]2+ + CH4 reaction a general consensus was reached concerning the methane oxidation mechanism, where the rate-limiting step involves homolytic C–H bond cleavage to form [Cu(OH)Cu]2+ with a physisorbed •CH3. An alternative possibility, i.e. heterolytic H-abstraction passing through a four-center transition state to give an intermediate with a Cu–CH3 bond, was given consideration only in a few recent DFT studies, but was found less favorable than radical C–H activation. In this contribution methane-to-methanol conversion by Cu–CHA is investigated using large cluster models and employing either DFT, with an extensive list of 97 functionals, or the high-level correlated DMRG/cu(4)-CASPT2 method. In all cases homolytic C–H dissociation most favorably proceeds via a (S = 1) transition state TS1r, whereas the transition state of heterolytic H-abstraction, TS1n, has an (S = 0) ground state. The DMRG/cu(4)-CASPT2 results convincingly point to the heterolytic route, with a calculated activation enthalpy of 12.3 kcal/mol, as compared to 21.1 kcal/mol for homolytic C–H dissociation. In contrast, the results obtained with DFT are strongly functional dependent. Conform with previous DFT studies, homolytic H-abstraction is preferred by the B3LYP functional (almost exclusively used in previous cluster model studies). However, many other functionals, hybrid meta-GGA functionals in particular, agree with DMRG/cu(4)-CASPT2 on heterolytic C–H activation. The present results reopen the debate on the general validity of the radical rebound mechanism for methane hydroxylation by a [CuOCu]2+ core in copper zeolites and also highlight the need for caution when relying on a specific DFT functional to elucidate oxidation reaction mechanisms in metal-based catalytic systems.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
