Chengyu Li, Zhaojun Liu, Chen Fu, Hongmin Li, Tong He, Gang Wu, Yanan Sheng, Ming Shen, Honglin Liu
{"title":"Hypoxia-induced degradation of FTO promotes apoptosis by unmasking RACK1-mediated activation of MTK1-JNK1/2 pathway","authors":"Chengyu Li, Zhaojun Liu, Chen Fu, Hongmin Li, Tong He, Gang Wu, Yanan Sheng, Ming Shen, Honglin Liu","doi":"10.1016/j.jare.2025.01.019","DOIUrl":null,"url":null,"abstract":"<div><h3>Introduction</h3><div>Hypoxia, a condition characterized by inadequate oxygen supply to tissues, triggers various cellular responses, including apoptosis. The RNA demethylase FTO has been shown to exert anti-apoptotic effects, but its functions independent of RNA demethylase—particularly those involving protein–protein interactions—during hypoxia remain unclear.</div></div><div><h3>Objectives</h3><div>This study aimed to elucidate the cytoprotective mechanism of FTO in preventing apoptosis under hypoxic stress.</div></div><div><h3>Methods</h3><div>NIH/3T3 cells, MEF cells, and mouse granulosa cells were cultured under hypoxia (1 % O<sub>2</sub>) and treated with inhibitors (chloroquine, MG132, cycloheximide) to identify FTO degradation pathways. RNA interference was used to knock down <em>atg7</em>, <em>nedd4</em>, and <em>fto</em>. Mass spectrometry identified FTO-associated proteins, and their interactions with FTO were analyzed with immunoprecipitation assays. FTO localization was examined through nuclear and cytoplasmic fractionation and fluorescence microscopy. Apoptosis was evaluated by flow cytometry (annexin V/PI). The role of FTO independent of its m6A demethylase activity was determined by inhibiting FTO function using FB23-2 or an H228A/D230A mutant lacking m6A demethylase activity.</div></div><div><h3>Results</h3><div>Upon hypoxia exposure, FTO relocated from the nucleus to the cytoplasm and underwent degradation through a regulatory pathway in which the E1-like ubiquitin-activating enzyme ATG7 and the E3 ubiquitin ligase NEDD4 cooperatively activated both the ubiquitin–proteasome system (UPS) and the autophagic-lysosomal pathway (ALP) in NIH/3T3 cells, MEF cells, and mouse granulosa cells. Furthermore, knocking down <em>atg7</em> resulted in FTO accumulation in the cytoplasm, where FTO exerted its protective effect by binding with RACK1, which impairs the interaction between RACK1 and MTK1, thereby blocking activation of JNK1/2 and subsequently preventing apoptosis in hypoxic cells.</div></div><div><h3>Conclusion</h3><div>This study reveals a novel function of cytoplasmic FTO in disrupting the RACK1-MTK1-JNK1/2-apoptosis cascade during hypoxia, positioning the functional context of FTO at the layer of protein–protein interactions, which extends its mechanistic role beyond RNA demethylation.</div></div>","PeriodicalId":14952,"journal":{"name":"Journal of Advanced Research","volume":"77 ","pages":"Pages 137-153"},"PeriodicalIF":13.0000,"publicationDate":"2025-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Advanced Research","FirstCategoryId":"103","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2090123225000384","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction
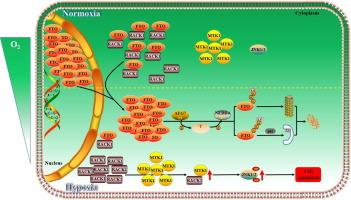
Hypoxia, a condition characterized by inadequate oxygen supply to tissues, triggers various cellular responses, including apoptosis. The RNA demethylase FTO has been shown to exert anti-apoptotic effects, but its functions independent of RNA demethylase—particularly those involving protein–protein interactions—during hypoxia remain unclear.
Objectives
This study aimed to elucidate the cytoprotective mechanism of FTO in preventing apoptosis under hypoxic stress.
Methods
NIH/3T3 cells, MEF cells, and mouse granulosa cells were cultured under hypoxia (1 % O2) and treated with inhibitors (chloroquine, MG132, cycloheximide) to identify FTO degradation pathways. RNA interference was used to knock down atg7, nedd4, and fto. Mass spectrometry identified FTO-associated proteins, and their interactions with FTO were analyzed with immunoprecipitation assays. FTO localization was examined through nuclear and cytoplasmic fractionation and fluorescence microscopy. Apoptosis was evaluated by flow cytometry (annexin V/PI). The role of FTO independent of its m6A demethylase activity was determined by inhibiting FTO function using FB23-2 or an H228A/D230A mutant lacking m6A demethylase activity.
Results
Upon hypoxia exposure, FTO relocated from the nucleus to the cytoplasm and underwent degradation through a regulatory pathway in which the E1-like ubiquitin-activating enzyme ATG7 and the E3 ubiquitin ligase NEDD4 cooperatively activated both the ubiquitin–proteasome system (UPS) and the autophagic-lysosomal pathway (ALP) in NIH/3T3 cells, MEF cells, and mouse granulosa cells. Furthermore, knocking down atg7 resulted in FTO accumulation in the cytoplasm, where FTO exerted its protective effect by binding with RACK1, which impairs the interaction between RACK1 and MTK1, thereby blocking activation of JNK1/2 and subsequently preventing apoptosis in hypoxic cells.
Conclusion
This study reveals a novel function of cytoplasmic FTO in disrupting the RACK1-MTK1-JNK1/2-apoptosis cascade during hypoxia, positioning the functional context of FTO at the layer of protein–protein interactions, which extends its mechanistic role beyond RNA demethylation.
期刊介绍:
Journal of Advanced Research (J. Adv. Res.) is an applied/natural sciences, peer-reviewed journal that focuses on interdisciplinary research. The journal aims to contribute to applied research and knowledge worldwide through the publication of original and high-quality research articles in the fields of Medicine, Pharmaceutical Sciences, Dentistry, Physical Therapy, Veterinary Medicine, and Basic and Biological Sciences.
The following abstracting and indexing services cover the Journal of Advanced Research: PubMed/Medline, Essential Science Indicators, Web of Science, Scopus, PubMed Central, PubMed, Science Citation Index Expanded, Directory of Open Access Journals (DOAJ), and INSPEC.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
