Lucca Blois, Renaldo T. Moura Jr, Ricardo L. Longo, Oscar L. Malta, Hermi F. Brito, Albano N. Carneiro Neto
{"title":"Assessment of DFT Functionals for Structural Determination of Lanthanide(III) Complexes Using Ligand Field Splitting","authors":"Lucca Blois, Renaldo T. Moura Jr, Ricardo L. Longo, Oscar L. Malta, Hermi F. Brito, Albano N. Carneiro Neto","doi":"10.1002/jcc.70034","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Lanthanide (Ln<sup>3+</sup>) tetrakis complexes, C[Ln(L)<sub>4</sub>], are important for applications due to their high quantum yields, solubility, and stability. Their luminescent properties depend on the structure, particularly the coordination polyhedron, the assessment of computational methods for calculating their structures is paramount. Usually, this assessment uses the RMSD of distances in the [Ln(L)<sub>4</sub>]<sup>−</sup> complex or {LnO<sub>8</sub>} polyhedron between crystallographic and calculated structures. However, since ligand field (LF) splitting is highly geometry-dependent, the RMSD between experimental LF splitting and Stark levels (RMSD-LF) offers a more accurate measure for evaluating quantum methods. LF energy eigenvalues were calculated using the simple overlap model (SOM), with geometries optimized by various density functionals. M06 and M06-L functionals, with def2-SVP/MWB52(Eu)/CPCM, demonstrate the best balance in best accuracy and low computational cost, making them suitable for modeling C[Eu(L)<sub>4</sub>] complexes.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 2","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70034","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
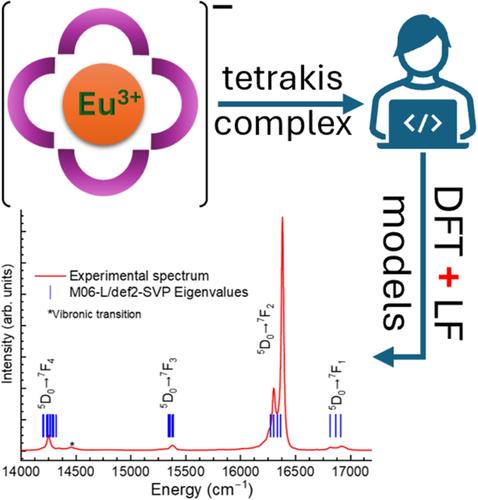
Lanthanide (Ln3+) tetrakis complexes, C[Ln(L)4], are important for applications due to their high quantum yields, solubility, and stability. Their luminescent properties depend on the structure, particularly the coordination polyhedron, the assessment of computational methods for calculating their structures is paramount. Usually, this assessment uses the RMSD of distances in the [Ln(L)4]− complex or {LnO8} polyhedron between crystallographic and calculated structures. However, since ligand field (LF) splitting is highly geometry-dependent, the RMSD between experimental LF splitting and Stark levels (RMSD-LF) offers a more accurate measure for evaluating quantum methods. LF energy eigenvalues were calculated using the simple overlap model (SOM), with geometries optimized by various density functionals. M06 and M06-L functionals, with def2-SVP/MWB52(Eu)/CPCM, demonstrate the best balance in best accuracy and low computational cost, making them suitable for modeling C[Eu(L)4] complexes.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
