{"title":"Machine Learning Committee Neural Network Potential Energy Surfaces for Two-Dimensional Metal–Organic Frameworks","authors":"Yuliang Shi, Farnaz A. Shakib","doi":"10.1021/acs.jpcc.4c07386","DOIUrl":null,"url":null,"abstract":"Two-dimensional (2D) layered metal–organic frameworks (MOFs) are gaining attention due to their unique structural and electronic properties with promising applications in compact electronic device fabrication. Long-time and large-scale molecular dynamics simulations of these materials can enhance and expedite the mapping out of their structure–property–function relationships for these applications. To make such simulations more feasible, herein, we construct a high-dimensional committee neural network potential (CNNP) for archetypal 2D MOFs Ni<sub>3</sub>(HIB)<sub>2</sub> and Ni<sub>3</sub>(HITP)<sub>2</sub> where HIB = hexaiminobenzene and HITP = hexaiminotriphenylene. We harness the power of active learning and committee neural networks to obtain a CNNP model by using only hundreds of snapshots from ab initio molecular dynamics (AIMD) trajectories. The developed CNNP model allows for simulations of thousands of atoms over extended time scales, which is typically unfeasible with AIMD simulations while maintaining the accuracy of the reference data. Our stable MD simulations based on the developed CNNP model reveal the flexible nature of the studied 2D MOFs at room temperature, including puckered layers, as opposed to the planar ones from 0 K electronic structure calculations. Furthermore, our model demonstrates transferability between bulk and monolayers, as well as different organic linkers. As the first model of its kind, we show that the high-dimensional CNNP models could be a reliable and effective approach for future studies on 2D MOFs.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"37 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c07386","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
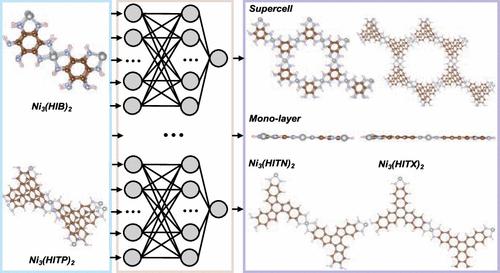
Two-dimensional (2D) layered metal–organic frameworks (MOFs) are gaining attention due to their unique structural and electronic properties with promising applications in compact electronic device fabrication. Long-time and large-scale molecular dynamics simulations of these materials can enhance and expedite the mapping out of their structure–property–function relationships for these applications. To make such simulations more feasible, herein, we construct a high-dimensional committee neural network potential (CNNP) for archetypal 2D MOFs Ni3(HIB)2 and Ni3(HITP)2 where HIB = hexaiminobenzene and HITP = hexaiminotriphenylene. We harness the power of active learning and committee neural networks to obtain a CNNP model by using only hundreds of snapshots from ab initio molecular dynamics (AIMD) trajectories. The developed CNNP model allows for simulations of thousands of atoms over extended time scales, which is typically unfeasible with AIMD simulations while maintaining the accuracy of the reference data. Our stable MD simulations based on the developed CNNP model reveal the flexible nature of the studied 2D MOFs at room temperature, including puckered layers, as opposed to the planar ones from 0 K electronic structure calculations. Furthermore, our model demonstrates transferability between bulk and monolayers, as well as different organic linkers. As the first model of its kind, we show that the high-dimensional CNNP models could be a reliable and effective approach for future studies on 2D MOFs.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
