{"title":"Scaled and Weighted Laplacian Matrices as Functional Descriptors for GPCR Ligands","authors":"Guillermo Goode-Romero, Laura Dominguez","doi":"10.1002/jcc.70015","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The G protein-coupled receptor (GPCR) pharmacology accounts for a significant field in research, clinical studies, and therapeutics. Computer-aided drug discovery is an evolving suite of techniques and methodologies that facilitate accelerated progress in drug discovery and repositioning. However, the structure–activity relationships of molecules targeting GPCRs are highly challenging in many cases since slight structural modifications can lead to drastic changes in biological functionality. Numerous molecular descriptors have been described, many of which successfully characterize the structural and physicochemical features of drug sets. Nonetheless, elucidating the structure–functionality relationships over extensive sets of drugs with multiple structural variations and known biological activity remains challenging in various biological systems. This work presents novel topological descriptors using Laplacian matrices, weighted, and scaled by atomic mass and partial charges. We tested these descriptors on three sets of GPCR ligands: muscarinic, β-adrenergic, and δ-opioid receptor ligands, evaluating their potential as functional descriptors of these receptors.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 3","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70015","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
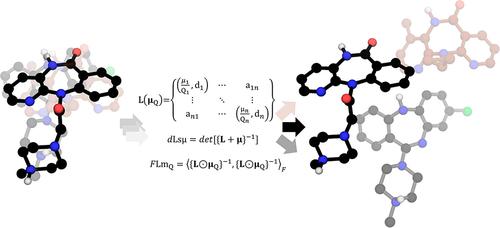
The G protein-coupled receptor (GPCR) pharmacology accounts for a significant field in research, clinical studies, and therapeutics. Computer-aided drug discovery is an evolving suite of techniques and methodologies that facilitate accelerated progress in drug discovery and repositioning. However, the structure–activity relationships of molecules targeting GPCRs are highly challenging in many cases since slight structural modifications can lead to drastic changes in biological functionality. Numerous molecular descriptors have been described, many of which successfully characterize the structural and physicochemical features of drug sets. Nonetheless, elucidating the structure–functionality relationships over extensive sets of drugs with multiple structural variations and known biological activity remains challenging in various biological systems. This work presents novel topological descriptors using Laplacian matrices, weighted, and scaled by atomic mass and partial charges. We tested these descriptors on three sets of GPCR ligands: muscarinic, β-adrenergic, and δ-opioid receptor ligands, evaluating their potential as functional descriptors of these receptors.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
