Alba Berzal-Serrano, Belén García-Bohórquez, Elena Aller, Teresa Jaijo, Inmaculada Pitarch-Castellano, Dolores Rausell, Gema García-García, José M Millán
{"title":"Outcomes of a Pilot Newborn Screening Program for Spinal Muscular Atrophy in the Valencian Community.","authors":"Alba Berzal-Serrano, Belén García-Bohórquez, Elena Aller, Teresa Jaijo, Inmaculada Pitarch-Castellano, Dolores Rausell, Gema García-García, José M Millán","doi":"10.3390/ijns11010007","DOIUrl":null,"url":null,"abstract":"<p><p>Spinal muscular atrophy (SMA) is a degenerative neuromuscular condition resulting from a homozygous deletion of the survival motor neuron 1 (<i>SMN1</i>) gene in 95% of patients. A timely diagnosis via newborn screening (NBS) and initiating treatment before the onset of symptoms are critical for improving health outcomes in affected individuals. We carried out a screening test by quantitative PCR (qPCR) to amplify the exon seven of <i>SMN1</i> using dried blood spot (DBS) samples. From October 2021 to August 2024, a total of 31,560 samples were tested in the Valencian Community (Spain) and 4 of them were positive for SMA, indicating an incidence of 1/7890. Genetic confirmation was performed using multiplex ligation-dependent probe amplification (MLPA) and AmplideX PCR/CE <i>SMN1/2</i> Plus kit, in parallel obtaining concordant results in survival motor neuron 2 (<i>SMN2</i>) gene copy number. Within the first few weeks of their lives, two of the four patients detected by NBS showed signs of severe hypotonia, becoming ineligible for treatment. The other two patients were the first presymptomatic patients with two copies of <i>SMN2</i> to receive treatment with Risdiplam in Spain. In order to treat positive cases in their early stages, we conclude that the official deployment of SMA newborn screening is necessary.</p>","PeriodicalId":14159,"journal":{"name":"International Journal of Neonatal Screening","volume":"11 1","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2025-01-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11755645/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Neonatal Screening","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ijns11010007","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
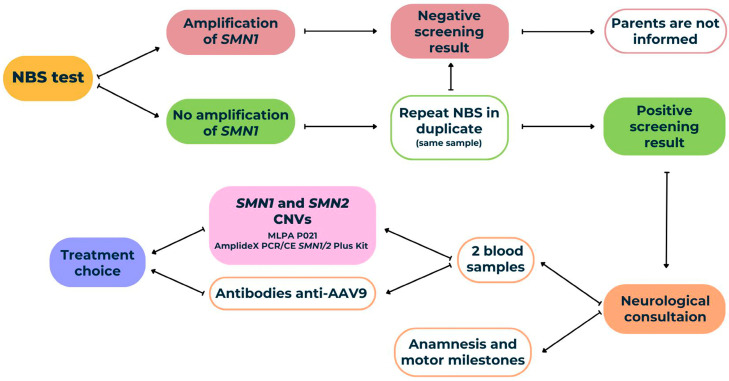
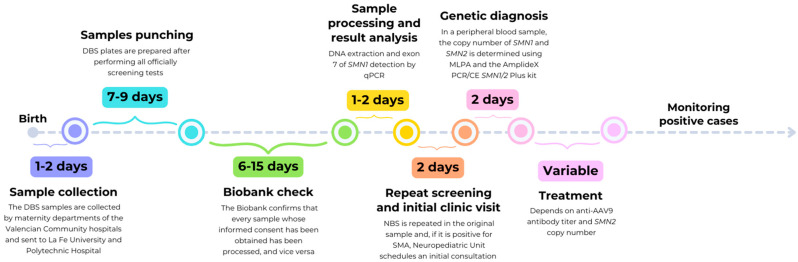
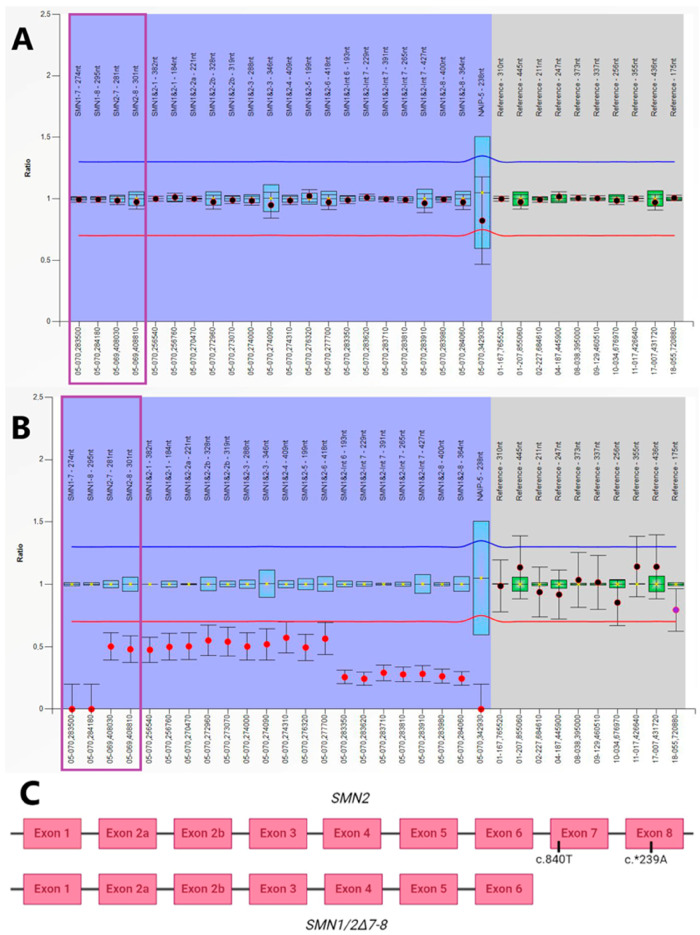
Spinal muscular atrophy (SMA) is a degenerative neuromuscular condition resulting from a homozygous deletion of the survival motor neuron 1 (SMN1) gene in 95% of patients. A timely diagnosis via newborn screening (NBS) and initiating treatment before the onset of symptoms are critical for improving health outcomes in affected individuals. We carried out a screening test by quantitative PCR (qPCR) to amplify the exon seven of SMN1 using dried blood spot (DBS) samples. From October 2021 to August 2024, a total of 31,560 samples were tested in the Valencian Community (Spain) and 4 of them were positive for SMA, indicating an incidence of 1/7890. Genetic confirmation was performed using multiplex ligation-dependent probe amplification (MLPA) and AmplideX PCR/CE SMN1/2 Plus kit, in parallel obtaining concordant results in survival motor neuron 2 (SMN2) gene copy number. Within the first few weeks of their lives, two of the four patients detected by NBS showed signs of severe hypotonia, becoming ineligible for treatment. The other two patients were the first presymptomatic patients with two copies of SMN2 to receive treatment with Risdiplam in Spain. In order to treat positive cases in their early stages, we conclude that the official deployment of SMA newborn screening is necessary.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
