{"title":"Lysosomes finely control macrophage inflammatory function via regulating the release of lysosomal Fe2+ through TRPML1 channel","authors":"Yanhong Xing, Meng-meng Wang, Feifei Zhang, Tianli Xin, Xinyan Wang, Rong Chen, Zhongheng Sui, Yawei Dong, Dongxue Xu, Xingyu Qian, Qixia Lu, Qingqing Li, Weijie Cai, Meiqin Hu, Yuqing Wang, Jun-li Cao, Derong Cui, Jiansong Qi, Wuyang Wang","doi":"10.1038/s41467-025-56403-x","DOIUrl":null,"url":null,"abstract":"<p>Lysosomes are best known for their roles in inflammatory responses by engaging in autophagy to remove inflammasomes. Here, we describe an unrecognized role for the lysosome, showing that it finely controls macrophage inflammatory function by manipulating the lysosomal Fe<sup>2+</sup>—prolyl hydroxylase domain enzymes (PHDs)—NF-κB—interleukin 1 beta (<i>IL1B</i>) transcription pathway that directly links lysosomes with inflammatory responses. TRPML1, a lysosomal cationic channel, is activated secondarily to ROS elevation upon inflammatory stimuli, which in turn suppresses <i>IL1B</i> transcription, thus limiting the excessive production of IL-1β in macrophages. Mechanistically, the suppression of <i>IL1B</i> transcription caused by TRPML1 activation results from its modulation on the release of lysosomal Fe<sup>2+</sup>, which subsequently activates PHDs. The activated PHDs then represses transcriptional activity of NF-κB, ultimately resulting in suppressed <i>IL1B</i> transcription. More importantly, in vivo stimulation of TRPML1 ameliorates multiple clinical signs of Dextran sulfate sodium-induced colitis in mice, suggesting TRPML1 has potential in treating inflammatory bowel disease.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"74 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-01-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-56403-x","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
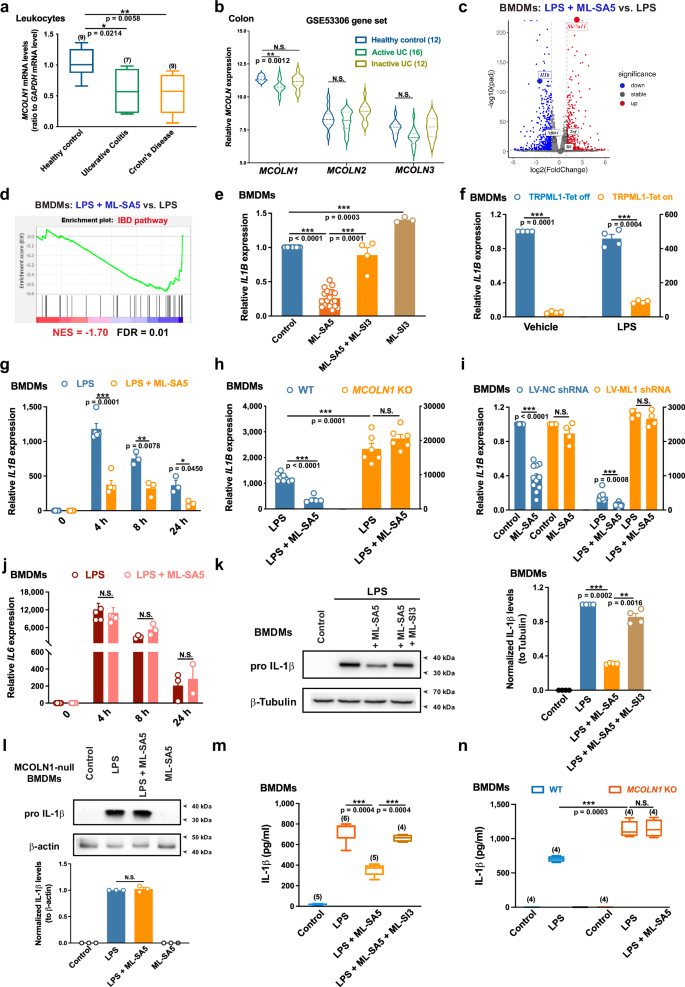
Lysosomes are best known for their roles in inflammatory responses by engaging in autophagy to remove inflammasomes. Here, we describe an unrecognized role for the lysosome, showing that it finely controls macrophage inflammatory function by manipulating the lysosomal Fe2+—prolyl hydroxylase domain enzymes (PHDs)—NF-κB—interleukin 1 beta (IL1B) transcription pathway that directly links lysosomes with inflammatory responses. TRPML1, a lysosomal cationic channel, is activated secondarily to ROS elevation upon inflammatory stimuli, which in turn suppresses IL1B transcription, thus limiting the excessive production of IL-1β in macrophages. Mechanistically, the suppression of IL1B transcription caused by TRPML1 activation results from its modulation on the release of lysosomal Fe2+, which subsequently activates PHDs. The activated PHDs then represses transcriptional activity of NF-κB, ultimately resulting in suppressed IL1B transcription. More importantly, in vivo stimulation of TRPML1 ameliorates multiple clinical signs of Dextran sulfate sodium-induced colitis in mice, suggesting TRPML1 has potential in treating inflammatory bowel disease.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
