Andrea Nedělníková, Petr Stadlbauer, Michal Otyepka, Petra Kührová, Markéta Paloncýová
{"title":"Atomistic Insights Into Interaction of Doxorubicin With DNA: From Duplex to Nucleosome","authors":"Andrea Nedělníková, Petr Stadlbauer, Michal Otyepka, Petra Kührová, Markéta Paloncýová","doi":"10.1002/jcc.70035","DOIUrl":null,"url":null,"abstract":"<p>Doxorubicin (DOX) is a widely used chemotherapeutic agent known for intercalating into DNA. However, the exact modes of DOX interactions with various DNA structures remain unclear. Using molecular dynamics (MD) simulations, we explored DOX interactions with DNA duplexes (dsDNA), G-quadruplex, and nucleosome. DOX predominantly stacks on terminal bases of dsDNA and occasionally binds into its minor groove. In the G-quadruplex, DOX stacks on planar tetrads but does not spontaneously intercalate into these structures. Potential of mean force calculations indicate that while intercalation is the most energetically favorable interaction mode for DOX in dsDNA, the process requires overcoming a significant energy barrier. In contrast, DOX spontaneously intercalates into bent nucleosomal DNA, due to the increased torsional stress. This preferential intercalation of DOX into regions with higher torsional stress provides new insights into its mechanism of action and underscores the importance of DNA tertiary and quaternary structures in therapies utilizing DNA intercalation.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 3","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70035","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70035","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
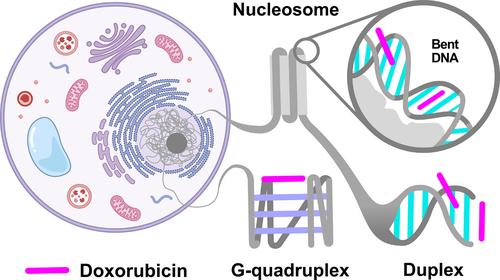
Doxorubicin (DOX) is a widely used chemotherapeutic agent known for intercalating into DNA. However, the exact modes of DOX interactions with various DNA structures remain unclear. Using molecular dynamics (MD) simulations, we explored DOX interactions with DNA duplexes (dsDNA), G-quadruplex, and nucleosome. DOX predominantly stacks on terminal bases of dsDNA and occasionally binds into its minor groove. In the G-quadruplex, DOX stacks on planar tetrads but does not spontaneously intercalate into these structures. Potential of mean force calculations indicate that while intercalation is the most energetically favorable interaction mode for DOX in dsDNA, the process requires overcoming a significant energy barrier. In contrast, DOX spontaneously intercalates into bent nucleosomal DNA, due to the increased torsional stress. This preferential intercalation of DOX into regions with higher torsional stress provides new insights into its mechanism of action and underscores the importance of DNA tertiary and quaternary structures in therapies utilizing DNA intercalation.
多柔比星(DOX)是一种广泛使用的化疗药物,以插入 DNA 而闻名。然而,DOX 与各种 DNA 结构相互作用的确切模式仍不清楚。利用分子动力学(MD)模拟,我们探索了 DOX 与 DNA 双链(dsDNA)、G-四链和核小体的相互作用。DOX 主要堆积在 dsDNA 的末端碱基上,偶尔也会结合到其小槽中。在 G 型四聚体中,DOX 堆叠在平面四聚体上,但不会自发地插入到这些结构中。平均力势计算表明,虽然插层是 DOX 在 dsDNA 中能量上最有利的相互作用模式,但这一过程需要克服巨大的能量障碍。相反,由于扭转应力增加,DOX 会自发地插层到弯曲的核糖体 DNA 中。DOX 优先插层到扭转应力较高的区域,这为了解 DOX 的作用机制提供了新的视角,并强调了 DNA 三级和四级结构在利用 DNA 插层疗法中的重要性。
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
