{"title":"First-principles elucidation of defect-mediated Li transport in hexagonal boron nitride†","authors":"Yilong Zhou, S. O. Kucheyev and Liwen F. Wan","doi":"10.1039/D4CP03655G","DOIUrl":null,"url":null,"abstract":"<p >Hexagonal boron nitride (hBN) is a promising candidate as a protective membrane or separator in Li-ion and Li–S batteries, given its excellent chemical stability, mechanical robustness, and high thermal conductivity. In addition, hBN can be functionalized by introducing defects and dopants, or be directly integrated into other active components of batteries, which further augments its appeal to the field. Here, we use first-principles simulations to evaluate the role of atomic defects in hBN in regulating the Li-ion diffusion mechanism and associated kinetics. Specifically, the following four distinct types of vacancy defects are considered: isolated single B and N vacancies, a B–N vacancy pair, and a B<small><sub>3</sub></small>N vacancy cluster. It is found that these defect sites generally favor Li intercalation and out-of-plane diffusion but slow down in-plane Li-ion diffusion due to a strong Li trapping effect at the defect sites. Such a trapping effect is, however, highly local such that it does not necessarily affect the overall Li-ion conductivity in defected hBN layers. The present systematic evaluation of the impact of atomic defects on Li ion migration and accompanied charge analysis of hBN lattice in response to Li-ion diffusion provide a mechanistic understanding of Li-ion transport behavior in defected hBN and highlight the potential of defect engineering to achieve optimal material performance.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 7","pages":" 3997-4003"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp03655g?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03655g","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
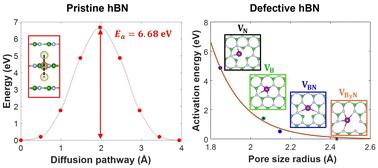
Hexagonal boron nitride (hBN) is a promising candidate as a protective membrane or separator in Li-ion and Li–S batteries, given its excellent chemical stability, mechanical robustness, and high thermal conductivity. In addition, hBN can be functionalized by introducing defects and dopants, or be directly integrated into other active components of batteries, which further augments its appeal to the field. Here, we use first-principles simulations to evaluate the role of atomic defects in hBN in regulating the Li-ion diffusion mechanism and associated kinetics. Specifically, the following four distinct types of vacancy defects are considered: isolated single B and N vacancies, a B–N vacancy pair, and a B3N vacancy cluster. It is found that these defect sites generally favor Li intercalation and out-of-plane diffusion but slow down in-plane Li-ion diffusion due to a strong Li trapping effect at the defect sites. Such a trapping effect is, however, highly local such that it does not necessarily affect the overall Li-ion conductivity in defected hBN layers. The present systematic evaluation of the impact of atomic defects on Li ion migration and accompanied charge analysis of hBN lattice in response to Li-ion diffusion provide a mechanistic understanding of Li-ion transport behavior in defected hBN and highlight the potential of defect engineering to achieve optimal material performance.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
