The Structural, Electronic and Vibrational Properties of LaCrO
3
$$ {}_3 $$
. A Quantum Mechanical Investigation by Using an All Electron Gaussian Type Basis Set and a Full Range Hybrid Functional
Khaled E. El-Kelany, Alexander Platonenko, Klaus Doll, Roberto Dovesi
{"title":"The Structural, Electronic and Vibrational Properties of LaCrO\n \n \n \n \n 3\n \n \n $$ {}_3 $$\n . A Quantum Mechanical Investigation by Using an All Electron Gaussian Type Basis Set and a Full Range Hybrid Functional","authors":"Khaled E. El-Kelany, Alexander Platonenko, Klaus Doll, Roberto Dovesi","doi":"10.1002/jcc.27523","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The geometrical, electronic and vibrational properties of LaCrO<span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mrow></mrow>\n <mn>3</mn>\n </msub>\n </mrow>\n <annotation>$$ {}_3 $$</annotation>\n </semantics></math> have been investigated by using an all electron Gaussian type basis set, the B3LYP functional and the CRYSTAL code, and compared with KVF<span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mrow></mrow>\n <mn>3</mn>\n </msub>\n </mrow>\n <annotation>$$ {}_3 $$</annotation>\n </semantics></math>: in the two compounds the transition metal is formally in d<span></span><math>\n <semantics>\n <mrow>\n <msup>\n <mrow></mrow>\n <mn>3</mn>\n </msup>\n </mrow>\n <annotation>$$ {}^3 $$</annotation>\n </semantics></math> configuration. The high spin t<span></span><math>\n <semantics>\n <mrow>\n <msubsup>\n <mrow></mrow>\n <mrow>\n <mn>2</mn>\n <mi>g</mi>\n </mrow>\n <mn>3</mn>\n </msubsup>\n </mrow>\n <annotation>$$ {}_{2g}^3 $$</annotation>\n </semantics></math> ground state excludes the Jahn Teller deformation and the orbital ordering. The energy gain due to the rotation of the octahedra (from the cubic space group Pm<span></span><math>\n <semantics>\n <mrow>\n <mover>\n <mn>3</mn>\n <mo>¯</mo>\n </mover>\n <mtext>m</mtext>\n </mrow>\n <annotation>$$ \\overline{3}\\mathrm{m} $$</annotation>\n </semantics></math>, N. 221, to space group <span></span><math>\n <semantics>\n <mrow>\n <mi>P</mi>\n <mfrac>\n <mn>4</mn>\n <mi>m</mi>\n </mfrac>\n <mi>bm</mi>\n </mrow>\n <annotation>$$ P\\frac{4}{m} bm $$</annotation>\n </semantics></math>, N.127, and to <span></span><math>\n <semantics>\n <mrow>\n <mi>I</mi>\n <mfrac>\n <mn>4</mn>\n <mi>m</mi>\n </mfrac>\n <mi>cm</mi>\n </mrow>\n <annotation>$$ I\\frac{4}{m} cm $$</annotation>\n </semantics></math>, N. 140) in the oxide is about 70 times larger than in the fluoride (5.4 vs. 0.08 mE<span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mrow></mrow>\n <mi>h</mi>\n </msub>\n </mrow>\n <annotation>$$ {}_h $$</annotation>\n </semantics></math>), due to the larger electrostatic forces (a factor four, as the formal charge doubles in going from F<sup>−</sup> to O<sup>2−</sup>) and the consequently reduced B-X distances. In KVF<span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mrow></mrow>\n <mn>3</mn>\n </msub>\n </mrow>\n <annotation>$$ {}_3 $$</annotation>\n </semantics></math>, the p states of fluorine are separated by 6.4 eV from the d states of vanadium, whose band is quite narrow (1 eV). In the oxide, on the contrary, the oxygen p states overlap to a large amount with the d states of chromium, whose band is more than 6 eV large. The FM and AFM energy differences, the spin density maps and profiles, and the Mulliken analysis data are also provided for documenting the differences between the oxide and the fluoride.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 4","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-02-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27523","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
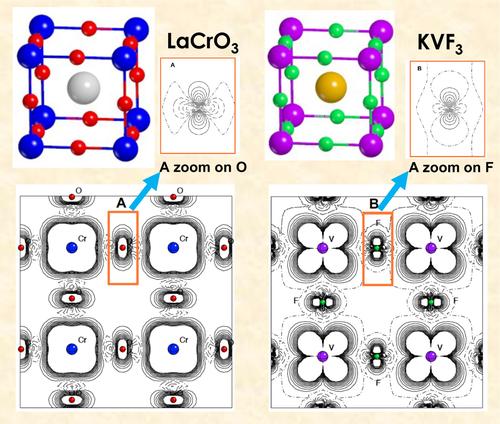
The geometrical, electronic and vibrational properties of LaCrO have been investigated by using an all electron Gaussian type basis set, the B3LYP functional and the CRYSTAL code, and compared with KVF: in the two compounds the transition metal is formally in d configuration. The high spin t ground state excludes the Jahn Teller deformation and the orbital ordering. The energy gain due to the rotation of the octahedra (from the cubic space group Pm, N. 221, to space group , N.127, and to , N. 140) in the oxide is about 70 times larger than in the fluoride (5.4 vs. 0.08 mE), due to the larger electrostatic forces (a factor four, as the formal charge doubles in going from F− to O2−) and the consequently reduced B-X distances. In KVF, the p states of fluorine are separated by 6.4 eV from the d states of vanadium, whose band is quite narrow (1 eV). In the oxide, on the contrary, the oxygen p states overlap to a large amount with the d states of chromium, whose band is more than 6 eV large. The FM and AFM energy differences, the spin density maps and profiles, and the Mulliken analysis data are also provided for documenting the differences between the oxide and the fluoride.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
