{"title":"Molecular and Electronic Structure and Properties of the Single Benzene-Based Fluorophores Containing Guanidine Subunit","authors":"Zoran Glasovac, Davor Margetić, Ivana Antol","doi":"10.1002/jcc.70054","DOIUrl":null,"url":null,"abstract":"<p>The Gibbs energies of protonation (Δ<i>G</i><sub>p</sub>) and the basic photophysical properties for single-benzene fluorophores (SBFs) containing guanidine and/or amino subunits and the changes that occur upon protonation were modeled by the TDDFT approach. The calculated Δ<i>G</i><sub>p</sub> energies for amino SBFs in the S<sub>1</sub> state range from 985 to 1100 kJ mol<sup>−1</sup> which are below the values for guanidines. The protonation of the guanidine-SBFs induces a hypsochromic shift of the absorption and the emission maxima with the Stokes shift of > 100 nm in both cases. Isomerization through the ESIPT process is less probable than in amino-SBFs due to the unfavorable thermodynamics. Still, if it occurs, it leads to a strong red shift of the emission by > 150 nm. Aromaticity indices point to strong antiaromatic character of the examined guanidino-SBFs in the FC region which decreases upon geometrical relaxation and ESIPT. The excited state proton transfer occurs in guanidine-SBF/phenol complexes in the S<sub>1</sub> state, stabilizing CT states and fluorescence quenching.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 4","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70054","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70054","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
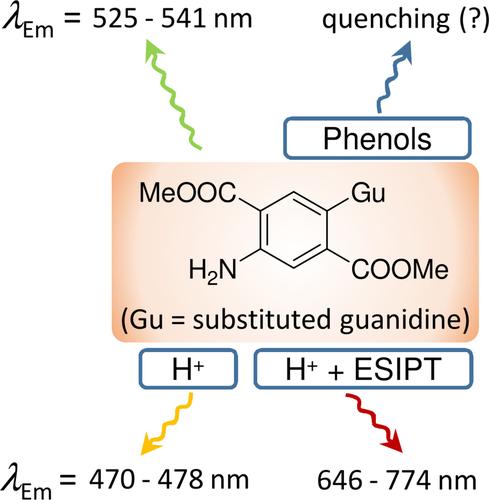
Abstract
The Gibbs energies of protonation (ΔGp) and the basic photophysical properties for single-benzene fluorophores (SBFs) containing guanidine and/or amino subunits and the changes that occur upon protonation were modeled by the TDDFT approach. The calculated ΔGp energies for amino SBFs in the S1 state range from 985 to 1100 kJ mol−1 which are below the values for guanidines. The protonation of the guanidine-SBFs induces a hypsochromic shift of the absorption and the emission maxima with the Stokes shift of > 100 nm in both cases. Isomerization through the ESIPT process is less probable than in amino-SBFs due to the unfavorable thermodynamics. Still, if it occurs, it leads to a strong red shift of the emission by > 150 nm. Aromaticity indices point to strong antiaromatic character of the examined guanidino-SBFs in the FC region which decreases upon geometrical relaxation and ESIPT. The excited state proton transfer occurs in guanidine-SBF/phenol complexes in the S1 state, stabilizing CT states and fluorescence quenching.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
