{"title":"Rules Describing CO2 Activation on Single-Atom Alloys from DFT-Meta-GGA Calculations and Artificial Intelligence","authors":"Herzain I. Rivera-Arrieta, Lucas Foppa","doi":"10.1021/acscatal.4c07178","DOIUrl":null,"url":null,"abstract":"Single-atom alloys (SAAs) arise as a promising concept for the design of improved CO<sub>2</sub> hydrogenation catalysts. However, from the immense number of possible SAA compositions and structures, only a few might display the properties required to be useful catalysts. Thus, the direct, high-throughput screening of materials is inefficient. Here, we use artificial intelligence to derive rules describing surface sites of SAAs that provide an effective CO<sub>2</sub> activation, a crucial initial step to convert the molecule into valuable products. We start by modeling the CO<sub>2</sub> interaction with 780 sites of flat and stepped surfaces of SAAs composed by Cu, Zn, and Pd hosts via high-quality DFT-mBEEF calculations. Then, we apply subgroup discovery to determine constraints on key physicochemical properties, out of 24 offered candidate descriptive parameters, characterizing subgroups (SGs) of surface sites where chemisorbed CO<sub>2</sub> displays large elongations of its C–O bonds. The key identified parameters are free-atom properties of the elements constituting the surface sites, such as their electron affinity, electronegativity, and radii of the d-orbitals. Additionally, the generalized coordination number is selected as a key geometrical parameter. The SG rules are applied to identify promising surface sites from a candidate space of over 1500 possible ones in different single-atom and dual-atom alloys. Some of the promising alloys predicted by the SG rules were explicitly tested by additional DFT-mBEEF calculations and confirmed to provide a significant CO<sub>2</sub> activation.","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"46 1","pages":""},"PeriodicalIF":13.1000,"publicationDate":"2025-02-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acscatal.4c07178","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
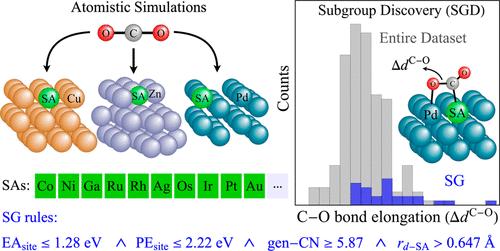
Single-atom alloys (SAAs) arise as a promising concept for the design of improved CO2 hydrogenation catalysts. However, from the immense number of possible SAA compositions and structures, only a few might display the properties required to be useful catalysts. Thus, the direct, high-throughput screening of materials is inefficient. Here, we use artificial intelligence to derive rules describing surface sites of SAAs that provide an effective CO2 activation, a crucial initial step to convert the molecule into valuable products. We start by modeling the CO2 interaction with 780 sites of flat and stepped surfaces of SAAs composed by Cu, Zn, and Pd hosts via high-quality DFT-mBEEF calculations. Then, we apply subgroup discovery to determine constraints on key physicochemical properties, out of 24 offered candidate descriptive parameters, characterizing subgroups (SGs) of surface sites where chemisorbed CO2 displays large elongations of its C–O bonds. The key identified parameters are free-atom properties of the elements constituting the surface sites, such as their electron affinity, electronegativity, and radii of the d-orbitals. Additionally, the generalized coordination number is selected as a key geometrical parameter. The SG rules are applied to identify promising surface sites from a candidate space of over 1500 possible ones in different single-atom and dual-atom alloys. Some of the promising alloys predicted by the SG rules were explicitly tested by additional DFT-mBEEF calculations and confirmed to provide a significant CO2 activation.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
