{"title":"SPAGRM: effectively controlling for sample relatedness in large-scale genome-wide association studies of longitudinal traits","authors":"He Xu, Yuzhuo Ma, Lin-lin Xu, Yin Li, Yufei Liu, Ying Li, Xu-jie Zhou, Wei Zhou, Seunggeun Lee, Peipei Zhang, Weihua Yue, Wenjian Bi","doi":"10.1038/s41467-025-56669-1","DOIUrl":null,"url":null,"abstract":"<p>Sample relatedness is a major confounder in genome-wide association studies (GWAS), potentially leading to inflated type I error rates if not appropriately controlled. A common strategy is to incorporate a random effect related to genetic relatedness matrix (GRM) into regression models. However, this approach is challenging for large-scale GWAS of complex traits, such as longitudinal traits. Here we propose a scalable and accurate analysis framework, SPA<sub>GRM</sub>, which controls for sample relatedness via a precise approximation of the joint distribution of genotypes. SPA<sub>GRM</sub> can utilize GRM-free models and thus is applicable to various trait types and statistical methods, including linear mixed models and generalized estimation equations for longitudinal traits. A hybrid strategy incorporating saddlepoint approximation greatly increases the accuracy to analyze low-frequency and rare genetic variants, especially in unbalanced phenotypic distributions. We also introduce SPA<sub>GRM(CCT)</sub> to aggregate the results following different models via Cauchy combination test. Extensive simulations and real data analyses demonstrated that SPA<sub>GRM</sub> maintains well-controlled type I error rates and SPA<sub>GRM(CCT)</sub> can serve as a broadly effective method. Applying SPA<sub>GRM</sub> to 79 longitudinal traits extracted from UK Biobank primary care data, we identified 7,463 genetic loci, making a pioneering attempt to conduct GWAS for these traits as longitudinal traits.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"21 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-56669-1","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
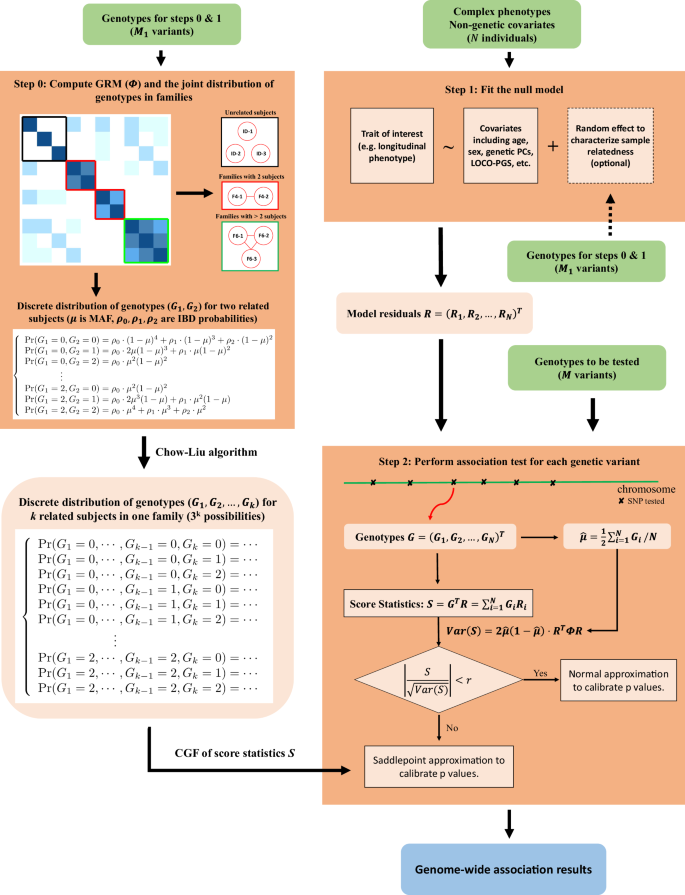
Sample relatedness is a major confounder in genome-wide association studies (GWAS), potentially leading to inflated type I error rates if not appropriately controlled. A common strategy is to incorporate a random effect related to genetic relatedness matrix (GRM) into regression models. However, this approach is challenging for large-scale GWAS of complex traits, such as longitudinal traits. Here we propose a scalable and accurate analysis framework, SPAGRM, which controls for sample relatedness via a precise approximation of the joint distribution of genotypes. SPAGRM can utilize GRM-free models and thus is applicable to various trait types and statistical methods, including linear mixed models and generalized estimation equations for longitudinal traits. A hybrid strategy incorporating saddlepoint approximation greatly increases the accuracy to analyze low-frequency and rare genetic variants, especially in unbalanced phenotypic distributions. We also introduce SPAGRM(CCT) to aggregate the results following different models via Cauchy combination test. Extensive simulations and real data analyses demonstrated that SPAGRM maintains well-controlled type I error rates and SPAGRM(CCT) can serve as a broadly effective method. Applying SPAGRM to 79 longitudinal traits extracted from UK Biobank primary care data, we identified 7,463 genetic loci, making a pioneering attempt to conduct GWAS for these traits as longitudinal traits.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
