A Novel Pathogenic Splicing Mutation of OFD1 is Responsible for a Boy with Joubert Syndrome Exhibiting Orofaciodigital Spectrum Anomalies, Polydactyly and Retinitis Pigmentosa.
Liang Chen, Mei-Fang Zhao, Hui-Wen Deng, Min Liao, Liang-Liang Fan, Qi-Bao Zhong, Jun Wang, Ke Li, Zheng-Hui Wu, Jian-Yin Yin
{"title":"A Novel Pathogenic Splicing Mutation of <i>OFD1</i> is Responsible for a Boy with Joubert Syndrome Exhibiting Orofaciodigital Spectrum Anomalies, Polydactyly and Retinitis Pigmentosa.","authors":"Liang Chen, Mei-Fang Zhao, Hui-Wen Deng, Min Liao, Liang-Liang Fan, Qi-Bao Zhong, Jun Wang, Ke Li, Zheng-Hui Wu, Jian-Yin Yin","doi":"10.2147/PGPM.S501623","DOIUrl":null,"url":null,"abstract":"<p><p>Joubert syndrome (JS) is an infrequent congenital neurodevelopmental ciliopathy, typically identified in children around the average age of 33 months. This disorder is characterized by developmental delay, cognitive impairment, and infantile hypotonia that may evolve into ataxia. Mutations in <i>OFD1</i> results in Joubert syndrome with a variety of phenotypes. Here, we identified a child who presented with Joubert syndrome exhibiting orofaciodigital spectrum anomalies, polydactyly, and retinitis pigmentosa. Whole exome sequencing and Sanger sequencing revealed a splicing mutation (NM_003611.2, c.2387+1G>A) in the <i>OFD1</i> gene of the patient and his mother. mRNA sequencing further confirmed this mutation. However, since the patient is homozygous and the mother is heterozygous, only the patient has the phenotype and the mother is normal. This mutation can lead to the loss of sixth coiled-coil domains of OFD1 protein, which further disrupt the ciliary signaling pathway and Hedgehog signaling pathway. This study presents a new case of JS and expands the mutant spectrum of <i>OFD1</i>, but also enhances our understanding of the mechanism by which <i>OFD1</i> is associated with ciliosis.</p>","PeriodicalId":56015,"journal":{"name":"Pharmacogenomics & Personalized Medicine","volume":"18 ","pages":"47-53"},"PeriodicalIF":1.8000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11804221/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pharmacogenomics & Personalized Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2147/PGPM.S501623","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
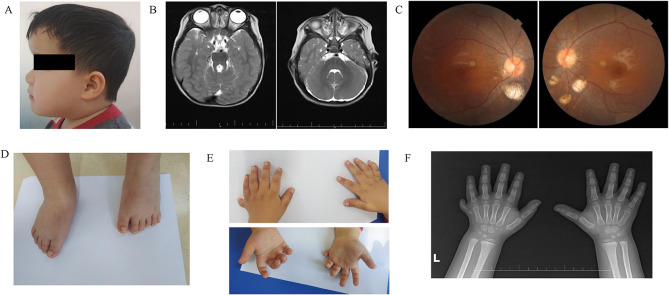
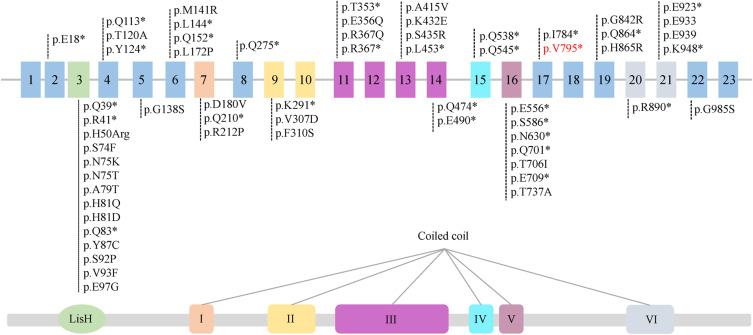
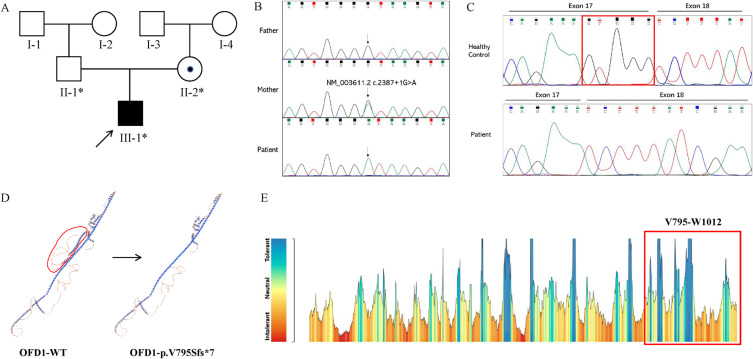
Joubert syndrome (JS) is an infrequent congenital neurodevelopmental ciliopathy, typically identified in children around the average age of 33 months. This disorder is characterized by developmental delay, cognitive impairment, and infantile hypotonia that may evolve into ataxia. Mutations in OFD1 results in Joubert syndrome with a variety of phenotypes. Here, we identified a child who presented with Joubert syndrome exhibiting orofaciodigital spectrum anomalies, polydactyly, and retinitis pigmentosa. Whole exome sequencing and Sanger sequencing revealed a splicing mutation (NM_003611.2, c.2387+1G>A) in the OFD1 gene of the patient and his mother. mRNA sequencing further confirmed this mutation. However, since the patient is homozygous and the mother is heterozygous, only the patient has the phenotype and the mother is normal. This mutation can lead to the loss of sixth coiled-coil domains of OFD1 protein, which further disrupt the ciliary signaling pathway and Hedgehog signaling pathway. This study presents a new case of JS and expands the mutant spectrum of OFD1, but also enhances our understanding of the mechanism by which OFD1 is associated with ciliosis.
期刊介绍:
Pharmacogenomics and Personalized Medicine is an international, peer-reviewed, open-access journal characterizing the influence of genotype on pharmacology leading to the development of personalized treatment programs and individualized drug selection for improved safety, efficacy and sustainability.
In particular, emphasis will be given to:
Genomic and proteomic profiling
Genetics and drug metabolism
Targeted drug identification and discovery
Optimizing drug selection & dosage based on patient''s genetic profile
Drug related morbidity & mortality intervention
Advanced disease screening and targeted therapeutic intervention
Genetic based vaccine development
Patient satisfaction and preference
Health economic evaluations
Practical and organizational issues in the development and implementation of personalized medicine programs.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
