Jaslyn Ru Ting Chen, Emily Xi Tan, Jingxiang Tang, Shi Xuan Leong, Sean Kai Xun Hue, Chi Seng Pun, In Yee Phang, Xing Yi Ling
{"title":"Machine Learning-Based SERS Chemical Space for Two-Way Prediction of Structures and Spectra of Untrained Molecules.","authors":"Jaslyn Ru Ting Chen, Emily Xi Tan, Jingxiang Tang, Shi Xuan Leong, Sean Kai Xun Hue, Chi Seng Pun, In Yee Phang, Xing Yi Ling","doi":"10.1021/jacs.4c15804","DOIUrl":null,"url":null,"abstract":"<p><p>Identifying unknown molecules beyond existing databases remains challenging in surface-enhanced Raman scattering (SERS) spectroscopy. Conventional SERS analysis relies on matching experimental and cataloged spectra, limiting identification to known molecules in databases. With a vast chemical space of >10<sup>60</sup> molecules, it is impractical to obtain the spectra of every molecule and rely solely on <i>in silico</i> techniques for spectral predictions. Here, we showcase an ML-based SERS chemical space that leverages key spectra-structure correlations to achieve two-way spectra-to-structure and structure-to-spectra predictions for untrained molecules with a >90% average accuracy. Using a SERS chemical space comprising 38 linear molecules from four classes (alcohols, aldehydes, amines, and carboxylic acids), our experimental and <i>in silico</i> studies reveal underlying spectral features that enable the prediction of untrained molecules represented by two molecular descriptors (functional group and carbon chain length). For forward spectra-to-structure predictions, we devise a two-step \"classification and regression\" ML framework to sequentially predict the functional group and carbon chain length of untrained molecules with 100% accuracy and ≤1 carbon difference, respectively. In addition, using an eXtreme Gradient Boosting (XGBoost) regressor trained on the two molecular descriptors, we attain inverse structure-to-spectra prediction with a high average cosine similarity of 90.4% between the predicted and experimental spectra. Our ML-based SERS chemical space represents a shift in molecular identification from traditional spectral matching to predictive modeling of spectra-structure relationships. These insights could motivate the expansion of SERS chemical spaces and realize demands for present and future SERS technologiesfor accurate unknown identification across diverse fields.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":" ","pages":"6654-6664"},"PeriodicalIF":15.6000,"publicationDate":"2025-02-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jacs.4c15804","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/14 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
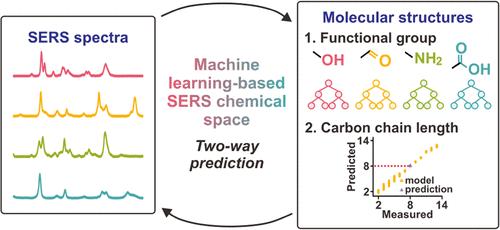
Identifying unknown molecules beyond existing databases remains challenging in surface-enhanced Raman scattering (SERS) spectroscopy. Conventional SERS analysis relies on matching experimental and cataloged spectra, limiting identification to known molecules in databases. With a vast chemical space of >1060 molecules, it is impractical to obtain the spectra of every molecule and rely solely on in silico techniques for spectral predictions. Here, we showcase an ML-based SERS chemical space that leverages key spectra-structure correlations to achieve two-way spectra-to-structure and structure-to-spectra predictions for untrained molecules with a >90% average accuracy. Using a SERS chemical space comprising 38 linear molecules from four classes (alcohols, aldehydes, amines, and carboxylic acids), our experimental and in silico studies reveal underlying spectral features that enable the prediction of untrained molecules represented by two molecular descriptors (functional group and carbon chain length). For forward spectra-to-structure predictions, we devise a two-step "classification and regression" ML framework to sequentially predict the functional group and carbon chain length of untrained molecules with 100% accuracy and ≤1 carbon difference, respectively. In addition, using an eXtreme Gradient Boosting (XGBoost) regressor trained on the two molecular descriptors, we attain inverse structure-to-spectra prediction with a high average cosine similarity of 90.4% between the predicted and experimental spectra. Our ML-based SERS chemical space represents a shift in molecular identification from traditional spectral matching to predictive modeling of spectra-structure relationships. These insights could motivate the expansion of SERS chemical spaces and realize demands for present and future SERS technologiesfor accurate unknown identification across diverse fields.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
