{"title":"High-Accuracy Long-Read Sequencing of <i>Mycobacterium tuberculosis</i> PSNK363 Isolated From the Democratic People's Republic of Korea.","authors":"Thi-Binh Dang, Nackmoon Sung, Kyunghyun Lim, Soyoung Lee, Jaehyun Jeon, Sanghoon Jheon","doi":"10.1155/cjid/2234550","DOIUrl":null,"url":null,"abstract":"<p><p>Long-read sequencing is a valuable technique for high-precision genome analysis. Despite the widespread use of the <i>Mycobacterium tuberculosis</i> H37Rv genome sequence as a reference for genetic variation analysis, its suitability for comparing clinical strains is limited. Therefore, we constructed the first known whole genome of a clinical <i>M. tuberculosis</i> strain, PSNK363, isolated from the Democratic People's Republic of Korea, using high-quality high-fidelity (HiFi) read sequencing and compared its genetic variations to those of H37Rv. PSNK363 was cultured to obtain genomic DNA, which was subjected to <i>de novo</i> whole-genome assembly using PacBio Sequel II with long-read HiFi sequencing. The sequences were compared to the reference genome H37Rv. HiFi long-read sequencing of <i>M. tuberculosis</i> PSNK363, with an accuracy of 99.99%, revealed a single circular chromosome of 4,422,110 bp, which is 10,578 bp longer than the H37Rv chromosome. The assembly had an average G + C content of 65.6%, 4079 protein-coding sequences, 53 tRNA genes, and 3 rRNA genes. Most genes (72.7%) were assigned as putative functions, whereas the remaining 27.3% were annotated as hypothetical. Comparison with H37Rv revealed a large inversion in the PSNK363 genome, which contains most of the deletion and insertion variants. <i>M. tuberculosis</i> PSNK363 had a longer genome sequence, more protein-coding genes, and a larger inversion region than H37Rv. High-accuracy whole-genome sequencing of PSNK363 holds the potential for enriching virulence databases and identifying informative loci for drug resistance analysis in <i>M. tuberculosis</i> isolates in the Democratic People's Republic of Korea.</p>","PeriodicalId":50715,"journal":{"name":"Canadian Journal of Infectious Diseases & Medical Microbiology","volume":"2025 ","pages":"2234550"},"PeriodicalIF":2.6000,"publicationDate":"2025-02-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11835475/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Canadian Journal of Infectious Diseases & Medical Microbiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1155/cjid/2234550","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"INFECTIOUS DISEASES","Score":null,"Total":0}
引用次数: 0
Abstract
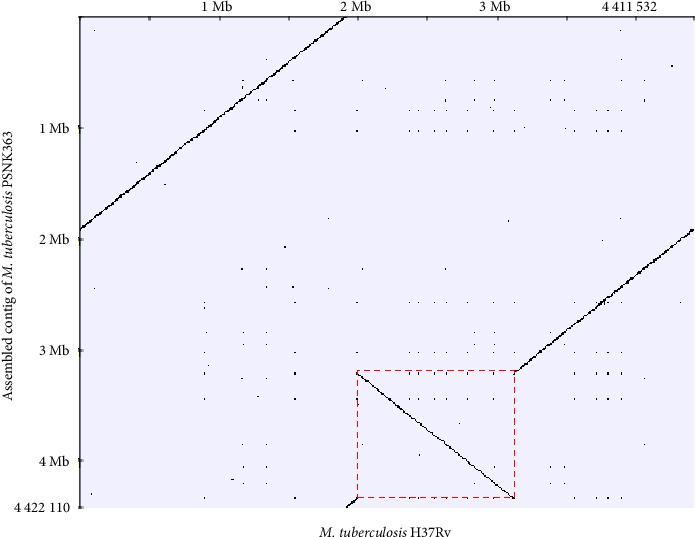
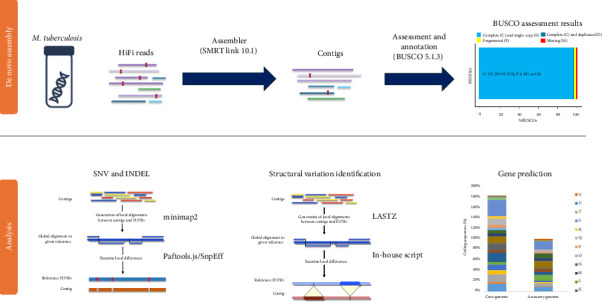
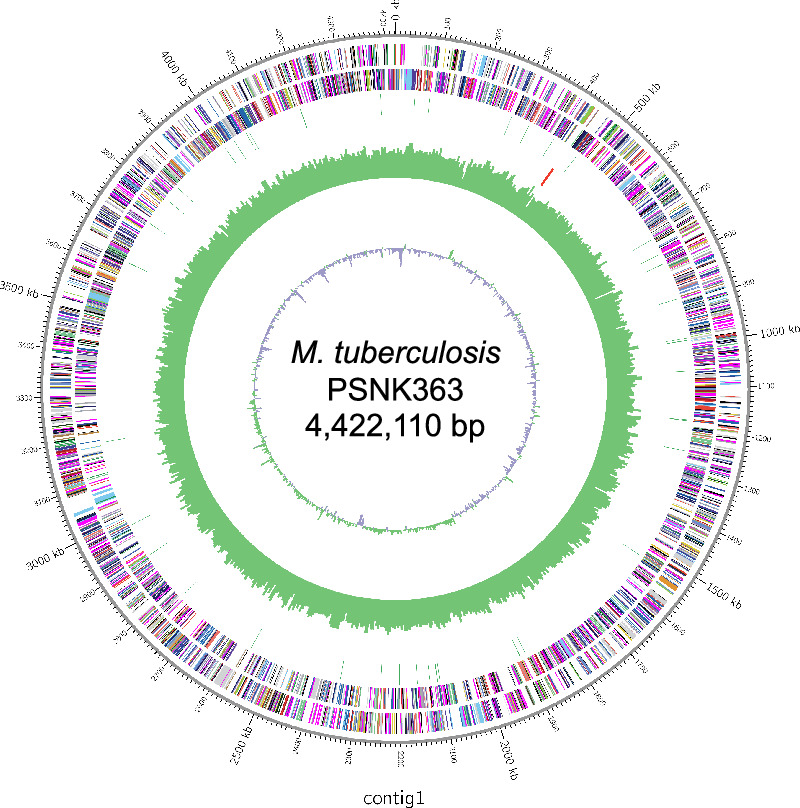
Long-read sequencing is a valuable technique for high-precision genome analysis. Despite the widespread use of the Mycobacterium tuberculosis H37Rv genome sequence as a reference for genetic variation analysis, its suitability for comparing clinical strains is limited. Therefore, we constructed the first known whole genome of a clinical M. tuberculosis strain, PSNK363, isolated from the Democratic People's Republic of Korea, using high-quality high-fidelity (HiFi) read sequencing and compared its genetic variations to those of H37Rv. PSNK363 was cultured to obtain genomic DNA, which was subjected to de novo whole-genome assembly using PacBio Sequel II with long-read HiFi sequencing. The sequences were compared to the reference genome H37Rv. HiFi long-read sequencing of M. tuberculosis PSNK363, with an accuracy of 99.99%, revealed a single circular chromosome of 4,422,110 bp, which is 10,578 bp longer than the H37Rv chromosome. The assembly had an average G + C content of 65.6%, 4079 protein-coding sequences, 53 tRNA genes, and 3 rRNA genes. Most genes (72.7%) were assigned as putative functions, whereas the remaining 27.3% were annotated as hypothetical. Comparison with H37Rv revealed a large inversion in the PSNK363 genome, which contains most of the deletion and insertion variants. M. tuberculosis PSNK363 had a longer genome sequence, more protein-coding genes, and a larger inversion region than H37Rv. High-accuracy whole-genome sequencing of PSNK363 holds the potential for enriching virulence databases and identifying informative loci for drug resistance analysis in M. tuberculosis isolates in the Democratic People's Republic of Korea.
期刊介绍:
Canadian Journal of Infectious Diseases and Medical Microbiology is a peer-reviewed, Open Access journal that publishes original research articles, review articles, and clinical studies related to infectious diseases of bacterial, viral and parasitic origin. The journal welcomes articles describing research on pathogenesis, epidemiology of infection, diagnosis and treatment, antibiotics and resistance, and immunology.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
