Jinxiao Sun, Dezhi Yang , Yongmi Huang, Zhihao Jiao, Shangzhe Yu, Yiru Liu, Kexin Gong, Guisen Zhao
{"title":"The discovery of novel N-heterocyclic-based AKT inhibitors with potential efficacy against prostate cancer","authors":"Jinxiao Sun, Dezhi Yang , Yongmi Huang, Zhihao Jiao, Shangzhe Yu, Yiru Liu, Kexin Gong, Guisen Zhao","doi":"10.1016/j.ejmech.2025.117435","DOIUrl":null,"url":null,"abstract":"<div><div>AKT, a serine/threonine protein kinase that plays a pivotal role in the PI3K/AKT/mTOR pathway, is overexpressed or hyperactivated in various cancers, including prostate, breast, and lung cancers. A series of novel nitrogen-containing aromatic heterocyclic compounds were designed, synthesized, and evaluated for AKT inhibition and anticancer activities. Among these, <strong>JL16</strong> and <strong>JL18</strong> emerged as potent inhibitors of AKT1 kinase, with IC<sub>50</sub> values of 7.1 ± 1.2 nM and 8.8 ± 1.3 nM, respectively. Both compounds also demonstrated significant antiproliferative effects against PC-3 prostate cancer cells, with IC<sub>50</sub> values of 2.9 ± 0.7 μM (<strong>JL16</strong>) and 3.0 ± 0.6 μM (<strong>JL18</strong>). Mechanistic studies revealed that <strong>JL16</strong> and <strong>JL18</strong> reduced phosphorylated GSK3β levels, confirming AKT target engagement in cells. Notably, <strong>JL18</strong> exhibited favorable pharmacokinetic properties in mice, including rapid oral absorption (T<sub>max</sub> = 0.5 h) and 41 % bioavailability. These findings highlight <strong>JL16</strong> and <strong>JL18</strong> as promising AKT inhibitors for further preclinical development.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"289 ","pages":"Article 117435"},"PeriodicalIF":5.9000,"publicationDate":"2025-05-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0223523425002004","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/23 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
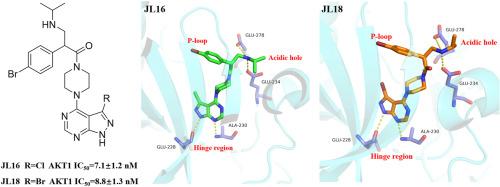
AKT, a serine/threonine protein kinase that plays a pivotal role in the PI3K/AKT/mTOR pathway, is overexpressed or hyperactivated in various cancers, including prostate, breast, and lung cancers. A series of novel nitrogen-containing aromatic heterocyclic compounds were designed, synthesized, and evaluated for AKT inhibition and anticancer activities. Among these, JL16 and JL18 emerged as potent inhibitors of AKT1 kinase, with IC50 values of 7.1 ± 1.2 nM and 8.8 ± 1.3 nM, respectively. Both compounds also demonstrated significant antiproliferative effects against PC-3 prostate cancer cells, with IC50 values of 2.9 ± 0.7 μM (JL16) and 3.0 ± 0.6 μM (JL18). Mechanistic studies revealed that JL16 and JL18 reduced phosphorylated GSK3β levels, confirming AKT target engagement in cells. Notably, JL18 exhibited favorable pharmacokinetic properties in mice, including rapid oral absorption (Tmax = 0.5 h) and 41 % bioavailability. These findings highlight JL16 and JL18 as promising AKT inhibitors for further preclinical development.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
