Tariq T. Ali , Madiha Merghani , Mohammed Al-Azzani , Luisa Maria Gatzemeier , Michael Hoppert , Dora Kaloyanova , Tiago F. Outeiro , Piotr Neumann , Blagovesta Popova , Gerhard H. Braus
{"title":"Rationally designed peptides inhibit the formation of α-synuclein fibrils and oligomers","authors":"Tariq T. Ali , Madiha Merghani , Mohammed Al-Azzani , Luisa Maria Gatzemeier , Michael Hoppert , Dora Kaloyanova , Tiago F. Outeiro , Piotr Neumann , Blagovesta Popova , Gerhard H. Braus","doi":"10.1016/j.ejmech.2025.117452","DOIUrl":null,"url":null,"abstract":"<div><div>Parkinson's Disease (PD) is characterized by the pathological aggregation of α-synuclein (αSyn) into oligomers and amyloid fibrils, making αSyn aggregation a key target for drug development. Peptides have gained recent attention as potential agents to inhibit aggregation. Two previously identified peptide inhibitors, discovered through large-scale yeast screening, were used as templates for <em>in silico</em> mutagenesis aimed at designing novel peptides with improved efficacy in inhibiting αSyn aggregation and cytotoxicity. The newly designed peptides underwent <em>in silico</em> docking analysis, and the most promising candidates were tested <em>in vitro</em> and in cellular models. Peptides T02 and T05 emerged as the most effective inhibitors, with T02 binding αSyn monomers and T05 targeting lower-order oligomers. Both peptides reduce αSyn fibril and oligomer formation <em>in vitro</em> and significantly suppress αSyn aggregation and cytotoxicity in yeast and human H4 cells. These novel peptides represent antagonists of αSyn aggregation with promising potential for therapeutic intervention for PD.</div></div>","PeriodicalId":314,"journal":{"name":"European Journal of Medicinal Chemistry","volume":"289 ","pages":"Article 117452"},"PeriodicalIF":5.9000,"publicationDate":"2025-05-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S022352342500217X","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/26 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
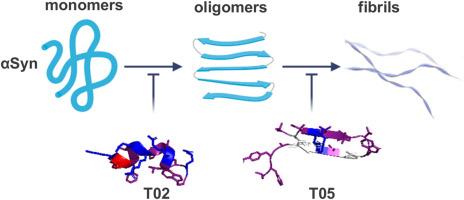
Parkinson's Disease (PD) is characterized by the pathological aggregation of α-synuclein (αSyn) into oligomers and amyloid fibrils, making αSyn aggregation a key target for drug development. Peptides have gained recent attention as potential agents to inhibit aggregation. Two previously identified peptide inhibitors, discovered through large-scale yeast screening, were used as templates for in silico mutagenesis aimed at designing novel peptides with improved efficacy in inhibiting αSyn aggregation and cytotoxicity. The newly designed peptides underwent in silico docking analysis, and the most promising candidates were tested in vitro and in cellular models. Peptides T02 and T05 emerged as the most effective inhibitors, with T02 binding αSyn monomers and T05 targeting lower-order oligomers. Both peptides reduce αSyn fibril and oligomer formation in vitro and significantly suppress αSyn aggregation and cytotoxicity in yeast and human H4 cells. These novel peptides represent antagonists of αSyn aggregation with promising potential for therapeutic intervention for PD.
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
