{"title":"Harnessing Pore Size in COF Membranes: A Concentration Gradient-Driven Molecular Dynamics Study on Enhanced H2/CH4 Separation","authors":"Parivash Jamshidi Ghaleh, Zeynep Pinar Haslak, Merdan Batyrow, Ilknur Erucar","doi":"10.1021/acsami.4c20420","DOIUrl":null,"url":null,"abstract":"This work presents a novel approach for accurately predicting the gas transport properties of covalent organic framework (COF) membranes using a nonequilibrium molecular dynamics (NEMD) methodology called concentration gradient-driven molecular dynamics (CGD-MD). We first simulated the flux of hydrogen (H<sub>2</sub>) and methane (CH<sub>4</sub>) across two distinct COF membranes, COF-300 and COF-320, for which experimental data are available in the literature. Our CGD-MD simulation results aligned closely with the experimentally measured gas permeability and selectivity of these COF membranes. Leveraging the same methodology, we discovered promising COF candidates for H<sub>2</sub>/CH<sub>4</sub> separation, including NPN-1, NPN-2, NPN-3, TPE-COF-I, COF-303, DMTA-TPB2, 3D-Por-COF, COF-921, COF-IM AA, TfpBDH, and PCOF-2. We then compared our findings with simulations utilizing the well-known approach that merges grand canonical Monte Carlo (GCMC) and equilibrium molecular dynamics (EMD) to predict gas adsorption and diffusion parameters in COFs. Our results showed that when the pore sizes of COF membranes are below 10 Å, the choice of the method plays a significant role in determining the performance of the membranes. The GCMC+EMD approach suggested that COFs tend to exhibit CH<sub>4</sub> selectivity when their pore limiting diameters are below 10 Å, whereas the CGD-MD results reveal a preference for H<sub>2</sub>. Density functional theory calculations indicate that H<sub>2</sub> has a lower affinity for three promising COFs, NPN-1, NPN-2, and NPN-3, compared to CH<sub>4</sub>, which results in H<sub>2</sub> remaining unbound, while CH<sub>4</sub> occupies all of the adsorption sites, thereby facilitating the selective recovery of H<sub>2</sub> at the end of the separation process. We proposed a relationship between adsorption time and diffusion time, highlighting the critical role of selecting an appropriate simulation method. This relationship underscores how adsorption and diffusion processes interplay, impacting material performance. Overall, these insights not only improve the accuracy of predictive models but also guide the development of more efficient COF-based membrane applications for future research and industrial applications.","PeriodicalId":5,"journal":{"name":"ACS Applied Materials & Interfaces","volume":"33 1","pages":""},"PeriodicalIF":8.2000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Materials & Interfaces","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1021/acsami.4c20420","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
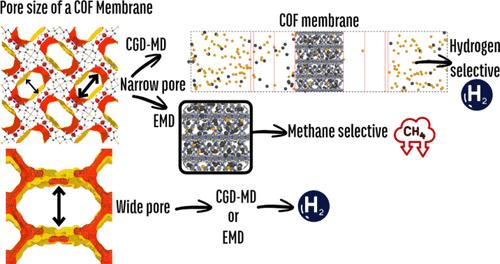
This work presents a novel approach for accurately predicting the gas transport properties of covalent organic framework (COF) membranes using a nonequilibrium molecular dynamics (NEMD) methodology called concentration gradient-driven molecular dynamics (CGD-MD). We first simulated the flux of hydrogen (H2) and methane (CH4) across two distinct COF membranes, COF-300 and COF-320, for which experimental data are available in the literature. Our CGD-MD simulation results aligned closely with the experimentally measured gas permeability and selectivity of these COF membranes. Leveraging the same methodology, we discovered promising COF candidates for H2/CH4 separation, including NPN-1, NPN-2, NPN-3, TPE-COF-I, COF-303, DMTA-TPB2, 3D-Por-COF, COF-921, COF-IM AA, TfpBDH, and PCOF-2. We then compared our findings with simulations utilizing the well-known approach that merges grand canonical Monte Carlo (GCMC) and equilibrium molecular dynamics (EMD) to predict gas adsorption and diffusion parameters in COFs. Our results showed that when the pore sizes of COF membranes are below 10 Å, the choice of the method plays a significant role in determining the performance of the membranes. The GCMC+EMD approach suggested that COFs tend to exhibit CH4 selectivity when their pore limiting diameters are below 10 Å, whereas the CGD-MD results reveal a preference for H2. Density functional theory calculations indicate that H2 has a lower affinity for three promising COFs, NPN-1, NPN-2, and NPN-3, compared to CH4, which results in H2 remaining unbound, while CH4 occupies all of the adsorption sites, thereby facilitating the selective recovery of H2 at the end of the separation process. We proposed a relationship between adsorption time and diffusion time, highlighting the critical role of selecting an appropriate simulation method. This relationship underscores how adsorption and diffusion processes interplay, impacting material performance. Overall, these insights not only improve the accuracy of predictive models but also guide the development of more efficient COF-based membrane applications for future research and industrial applications.
期刊介绍:
ACS Applied Materials & Interfaces is a leading interdisciplinary journal that brings together chemists, engineers, physicists, and biologists to explore the development and utilization of newly-discovered materials and interfacial processes for specific applications. Our journal has experienced remarkable growth since its establishment in 2009, both in terms of the number of articles published and the impact of the research showcased. We are proud to foster a truly global community, with the majority of published articles originating from outside the United States, reflecting the rapid growth of applied research worldwide.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
