{"title":"Correction to “Predicting and Understanding Noncovalent Interactions Using Novel Forms of Symmetry-Adapted Perturbation Theory”","authors":"Kevin Carter-Fenk, Ka Un Lao, John M. Herbert","doi":"10.1021/acs.accounts.5c00107","DOIUrl":null,"url":null,"abstract":"In a previous Account, (1) we surveyed the use of extended symmetry-adapted perturbation theory (XSAPT), a family of methods for computing accurate intermolecular interaction energies and components thereof. In considering π-stacking interactions, we made comparisons between the XSAPT + many-body dispersion (MBD) method and a model potential introduced in a seminal paper on π–π interactions by Hunter and Sanders (HS). (2) Unfortunately, our implementation of the HS model contained an error in the van der Waals (vdW) term, which is corrected here alongside some additional clarifications. Because there are subtleties in how the vdW parameters were originally reported, (2) as well as ambiguity regarding which point charges constitute the HS model, (2,3) additional details are provided here. The HS model consists of a point-charge electrostatic term (<i>E</i><sub>elst</sub><sup>Q</sup>) and a vdW term (<i>E</i><sub>vdW</sub>), There is some ambiguity regarding the point charges to be used in <i>E</i><sub>elst</sub><sup>Q</sup>. What is clear is that the HS model contains atom-centered point charges for carbon atoms within the π-system (<i>q</i><sub>C</sub>) along with out-of-plane displaced charges (<i>q</i><sub>π</sub>) to represent the π-electrons. In their original 1990 paper, HS first discuss “unpolarized” or “idealized” charges, in which carbon atoms within the π-system are described by charges <i>q</i><sub>C</sub> = +1.0 and <i>q</i><sub>π</sub> = −0.5 (in atomic units). (2) The π charges are displaced from the nuclei by δ = 0.47 Å, both above and below the arene plane, a value that is determined in order to reproduce the experimental quadrupole moment of C<sub>6</sub>H<sub>6</sub>. (5) Although the HS paper includes a discussion of polarizing this idealized framework, no actual values for hydrogen-atom charges are provided in ref (2). Moreover, Figure 3 of ref (2) depicts only <i>q</i><sub>C</sub> = +1.0 and <i>q</i><sub>π</sub> = −0.5, with no indication that there are charges on the hydrogen atoms. In 1991, Hunter et al. (3) suggested a model in which the charge on carbon is reduced to <i>q</i><sub>C</sub> = +0.95 and a charge <i>q</i><sub>H</sub> = +0.05 is placed on hydrogen, retaining <i>q</i><sub>π</sub> = −0.5. This scheme (in Figure 3 of ref (3)) is attributed to the original HS model even though the value of <i>q</i><sub>H</sub> was not provided in the original. In other work by Hunter and co-workers, only <i>q</i><sub>C</sub> and <i>q</i><sub>π</sub> are discussed, e.g., in Figure 3 of ref (6). These ambiguities are consistent with widespread confusion in the literature regarding what the HS model actually is, as discussed elsewhere. (7) For this Correction, we implemented <i>E</i><sub>elst</sub><sup>Q</sup> according to ref (3) using <i>q</i><sub>C</sub> = +0.95, <i>q</i><sub>H</sub> = +0.05, and <i>q</i><sub>π</sub> = −0.5. For (C<sub>6</sub>H<sub>6</sub>)<sub>2</sub>, the presence or absence of <i>q</i><sub>H</sub> makes only a minor difference. Using the corrected parameters in <i>E</i><sub>vdW</sub> and the updated parameters in <i>E</i><sub>elst</sub><sup>Q</sup>, we recomputed HS potentials for the lateral displacement of parallel and perpendicular arrangements of the benzene dimer. Figure 1 of this Correction should replace Figure 8 in ref (1), illustrating how <i>E</i><sub>HS</sub> and <i>E</i><sub>elst</sub><sup>Q</sup> vary with lateral displacement. Note that the electrostatic component (Figure 1b) remains qualitatively incorrect in comparison to a calculation based on full monomer charge densities, the latter of which can be found in Figure 9 of ref (1). The crux of our argument is unchanged, namely, that electrostatics does not explain parallel-displaced π-stacking. Figure 1. (a) Total HS model potential and (b) its electrostatic component, for lateral displacement of (C<sub>6</sub>H<sub>6</sub>)<sub>2</sub> in either a coplanar configuration at 3.4 Å separation (consistent with the parallel-displaced minimum-energy geometry) or else a perpendicular edge-to-face arrangement with a 5.0 Å center-to-center distance, consistent with the T-shaped saddle point of (C<sub>6</sub>H<sub>6</sub>)<sub>2</sub>. This figure should replace Figure 8 of ref (1). Figure 2 of this Correction plots <i>E</i><sub>HS</sub> and <i>E</i><sub>elst</sub><sup>Q</sup> for lateral displacement of benzene atop a C<sub>96</sub>H<sub>24</sub> graphene nanoflake; this figure should replace Figure 12a of ref (1). As compared to XSAPT + MBD calculations (in Figure 12c of ref (1)), the corrected version of the HS model potential is qualitatively correct insofar as the zero-displacement structure is a saddle point between symmetry-equivalent minima corresponding to parallel-displaced π-stacking. Nevertheless, the electrostatic component <i>E</i><sub>elst</sub><sup>Q</sup> (Figure 2b of this Correction) is repulsive at all values of the lateral displacement coordinate, which is not consistent with exact electrostatics. (8) Indeed, a central aspect of our “pizza-π” model of π–π interactions, (8) which explains how π-stacking is different from ordinary dispersion, is that electrostatics is attractive for two coplanar arenes separated by typical π-stacking distances of 3.4–3.8 Å. This conclusion is borne out in a wide variety of π–π interactions. (7−11) Figure 2. (a) Total HS model potential and (b) its electrostatic component, for lateral displacement of C<sub>6</sub>H<sub>6</sub> atop C<sub>96</sub>H<sub>24</sub> in a cofacial configuration at 3.4 Å separation. This figure should replace Figure 12a of ref (1). A final topic of discussion is the use of δ<sup>±</sup> to label the cartoons in Figure 6 of ref (1). This is potentially misleading, as it may suggest that C<sub>6</sub>F<sub>6</sub> has a positive charge density on its π faces, which is absurd. Rather, these diagrams are intended to convey the change in <i>quadrupolar</i> electrostatics that occurs when one C<sub>6</sub>H<sub>6</sub> monomer is replaced by C<sub>6</sub>F<sub>6</sub>, since the quadrupole–quadrupole interactions in (C<sub>6</sub>H<sub>6</sub>)···(C<sub>6</sub>F<sub>6</sub>) are attractive in the face-to-face orientation. A similar diagram, coloring C<sub>6</sub>F<sub>6</sub> opposite to C<sub>6</sub>H<sub>6</sub>, can be found in Hunter et al. (6) Although these cartoon charge distributions are widely used in discussing π-stacking, (6,12,13) we have elsewhere suggested that they are misleading and that their use ought to be discontinued. (7) We thank Prof. Steven Wheeler (University of Georgia) for bringing these issues to our attention based on his independent implementation of the HS model. This article references 13 other publications. This article has not yet been cited by other publications.","PeriodicalId":1,"journal":{"name":"Accounts of Chemical Research","volume":"49 1","pages":""},"PeriodicalIF":17.7000,"publicationDate":"2025-03-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Accounts of Chemical Research","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.accounts.5c00107","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
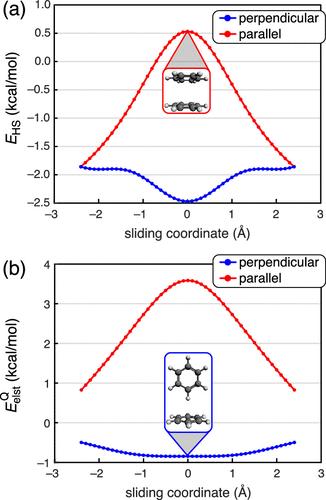
In a previous Account, (1) we surveyed the use of extended symmetry-adapted perturbation theory (XSAPT), a family of methods for computing accurate intermolecular interaction energies and components thereof. In considering π-stacking interactions, we made comparisons between the XSAPT + many-body dispersion (MBD) method and a model potential introduced in a seminal paper on π–π interactions by Hunter and Sanders (HS). (2) Unfortunately, our implementation of the HS model contained an error in the van der Waals (vdW) term, which is corrected here alongside some additional clarifications. Because there are subtleties in how the vdW parameters were originally reported, (2) as well as ambiguity regarding which point charges constitute the HS model, (2,3) additional details are provided here. The HS model consists of a point-charge electrostatic term (EelstQ) and a vdW term (EvdW), There is some ambiguity regarding the point charges to be used in EelstQ. What is clear is that the HS model contains atom-centered point charges for carbon atoms within the π-system (qC) along with out-of-plane displaced charges (qπ) to represent the π-electrons. In their original 1990 paper, HS first discuss “unpolarized” or “idealized” charges, in which carbon atoms within the π-system are described by charges qC = +1.0 and qπ = −0.5 (in atomic units). (2) The π charges are displaced from the nuclei by δ = 0.47 Å, both above and below the arene plane, a value that is determined in order to reproduce the experimental quadrupole moment of C6H6. (5) Although the HS paper includes a discussion of polarizing this idealized framework, no actual values for hydrogen-atom charges are provided in ref (2). Moreover, Figure 3 of ref (2) depicts only qC = +1.0 and qπ = −0.5, with no indication that there are charges on the hydrogen atoms. In 1991, Hunter et al. (3) suggested a model in which the charge on carbon is reduced to qC = +0.95 and a charge qH = +0.05 is placed on hydrogen, retaining qπ = −0.5. This scheme (in Figure 3 of ref (3)) is attributed to the original HS model even though the value of qH was not provided in the original. In other work by Hunter and co-workers, only qC and qπ are discussed, e.g., in Figure 3 of ref (6). These ambiguities are consistent with widespread confusion in the literature regarding what the HS model actually is, as discussed elsewhere. (7) For this Correction, we implemented EelstQ according to ref (3) using qC = +0.95, qH = +0.05, and qπ = −0.5. For (C6H6)2, the presence or absence of qH makes only a minor difference. Using the corrected parameters in EvdW and the updated parameters in EelstQ, we recomputed HS potentials for the lateral displacement of parallel and perpendicular arrangements of the benzene dimer. Figure 1 of this Correction should replace Figure 8 in ref (1), illustrating how EHS and EelstQ vary with lateral displacement. Note that the electrostatic component (Figure 1b) remains qualitatively incorrect in comparison to a calculation based on full monomer charge densities, the latter of which can be found in Figure 9 of ref (1). The crux of our argument is unchanged, namely, that electrostatics does not explain parallel-displaced π-stacking. Figure 1. (a) Total HS model potential and (b) its electrostatic component, for lateral displacement of (C6H6)2 in either a coplanar configuration at 3.4 Å separation (consistent with the parallel-displaced minimum-energy geometry) or else a perpendicular edge-to-face arrangement with a 5.0 Å center-to-center distance, consistent with the T-shaped saddle point of (C6H6)2. This figure should replace Figure 8 of ref (1). Figure 2 of this Correction plots EHS and EelstQ for lateral displacement of benzene atop a C96H24 graphene nanoflake; this figure should replace Figure 12a of ref (1). As compared to XSAPT + MBD calculations (in Figure 12c of ref (1)), the corrected version of the HS model potential is qualitatively correct insofar as the zero-displacement structure is a saddle point between symmetry-equivalent minima corresponding to parallel-displaced π-stacking. Nevertheless, the electrostatic component EelstQ (Figure 2b of this Correction) is repulsive at all values of the lateral displacement coordinate, which is not consistent with exact electrostatics. (8) Indeed, a central aspect of our “pizza-π” model of π–π interactions, (8) which explains how π-stacking is different from ordinary dispersion, is that electrostatics is attractive for two coplanar arenes separated by typical π-stacking distances of 3.4–3.8 Å. This conclusion is borne out in a wide variety of π–π interactions. (7−11) Figure 2. (a) Total HS model potential and (b) its electrostatic component, for lateral displacement of C6H6 atop C96H24 in a cofacial configuration at 3.4 Å separation. This figure should replace Figure 12a of ref (1). A final topic of discussion is the use of δ± to label the cartoons in Figure 6 of ref (1). This is potentially misleading, as it may suggest that C6F6 has a positive charge density on its π faces, which is absurd. Rather, these diagrams are intended to convey the change in quadrupolar electrostatics that occurs when one C6H6 monomer is replaced by C6F6, since the quadrupole–quadrupole interactions in (C6H6)···(C6F6) are attractive in the face-to-face orientation. A similar diagram, coloring C6F6 opposite to C6H6, can be found in Hunter et al. (6) Although these cartoon charge distributions are widely used in discussing π-stacking, (6,12,13) we have elsewhere suggested that they are misleading and that their use ought to be discontinued. (7) We thank Prof. Steven Wheeler (University of Georgia) for bringing these issues to our attention based on his independent implementation of the HS model. This article references 13 other publications. This article has not yet been cited by other publications.
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
