Yi Fu, Max Land, Tamar Kavlashvili, Ruobing Cui, Minsoo Kim, Emily DeBitetto, Toby Lieber, Keun Woo Ryu, Elim Choi, Ignas Masilionis, Rahul Saha, Meril Takizawa, Daphne Baker, Marco Tigano, Caleb A. Lareau, Ed Reznik, Roshan Sharma, Ronan Chaligne, Craig B. Thompson, Dana Pe’er, Agnel Sfeir
{"title":"Engineering mtDNA deletions by reconstituting end joining in human mitochondria","authors":"Yi Fu, Max Land, Tamar Kavlashvili, Ruobing Cui, Minsoo Kim, Emily DeBitetto, Toby Lieber, Keun Woo Ryu, Elim Choi, Ignas Masilionis, Rahul Saha, Meril Takizawa, Daphne Baker, Marco Tigano, Caleb A. Lareau, Ed Reznik, Roshan Sharma, Ronan Chaligne, Craig B. Thompson, Dana Pe’er, Agnel Sfeir","doi":"10.1016/j.cell.2025.02.009","DOIUrl":null,"url":null,"abstract":"Recent breakthroughs in the genetic manipulation of mitochondrial DNA (mtDNA) have enabled precise base substitutions and the efficient elimination of genomes carrying pathogenic mutations. However, reconstituting mtDNA deletions linked to mitochondrial myopathies remains challenging. Here, we engineered mtDNA deletions in human cells by co-expressing end-joining (EJ) machinery and targeted endonucleases. Using mitochondrial EJ (mito-EJ) and mito-ScaI, we generated a panel of clonal cell lines harboring a ∼3.5 kb mtDNA deletion across the full spectrum of heteroplasmy. Investigating these cells revealed a critical threshold of ∼75% deleted genomes, beyond which oxidative phosphorylation (OXPHOS) protein depletion, metabolic disruption, and impaired growth in galactose-containing media were observed. Single-cell multiomic profiling identified two distinct nuclear gene deregulation responses: one triggered at the deletion threshold and another progressively responding to heteroplasmy. Ultimately, we show that our method enables the modeling of disease-associated mtDNA deletions across cell types and could inform the development of targeted therapies.","PeriodicalId":9656,"journal":{"name":"Cell","volume":"91 1","pages":""},"PeriodicalIF":42.5000,"publicationDate":"2025-03-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.cell.2025.02.009","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
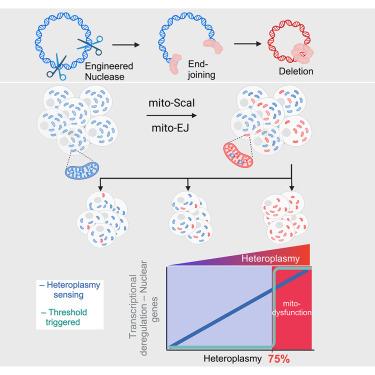
Recent breakthroughs in the genetic manipulation of mitochondrial DNA (mtDNA) have enabled precise base substitutions and the efficient elimination of genomes carrying pathogenic mutations. However, reconstituting mtDNA deletions linked to mitochondrial myopathies remains challenging. Here, we engineered mtDNA deletions in human cells by co-expressing end-joining (EJ) machinery and targeted endonucleases. Using mitochondrial EJ (mito-EJ) and mito-ScaI, we generated a panel of clonal cell lines harboring a ∼3.5 kb mtDNA deletion across the full spectrum of heteroplasmy. Investigating these cells revealed a critical threshold of ∼75% deleted genomes, beyond which oxidative phosphorylation (OXPHOS) protein depletion, metabolic disruption, and impaired growth in galactose-containing media were observed. Single-cell multiomic profiling identified two distinct nuclear gene deregulation responses: one triggered at the deletion threshold and another progressively responding to heteroplasmy. Ultimately, we show that our method enables the modeling of disease-associated mtDNA deletions across cell types and could inform the development of targeted therapies.
期刊介绍:
Cells is an international, peer-reviewed, open access journal that focuses on cell biology, molecular biology, and biophysics. It is affiliated with several societies, including the Spanish Society for Biochemistry and Molecular Biology (SEBBM), Nordic Autophagy Society (NAS), Spanish Society of Hematology and Hemotherapy (SEHH), and Society for Regenerative Medicine (Russian Federation) (RPO).
The journal publishes research findings of significant importance in various areas of experimental biology, such as cell biology, molecular biology, neuroscience, immunology, virology, microbiology, cancer, human genetics, systems biology, signaling, and disease mechanisms and therapeutics. The primary criterion for considering papers is whether the results contribute to significant conceptual advances or raise thought-provoking questions and hypotheses related to interesting and important biological inquiries.
In addition to primary research articles presented in four formats, Cells also features review and opinion articles in its "leading edge" section, discussing recent research advancements and topics of interest to its wide readership.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
