Laura Gómez-Dabó, Arnau Llaurado, Daniel Sánchez-Tejerina, Victoria González, Carmen Montalvo-Olmedo, Carlos Lázaro-Hernández, Marc Rodrigo-Gisbert, Samuel López-Maza, Maider Iza-Achutegui, Lídia Giramé-Rizzo, Nuria Raguer, Raúl Juntas
{"title":"A Rare Guillain-Barré Syndrome Variant with Multi-Ganglioside Reactivity: A Case of Severe Cranial Nerve Involvement.","authors":"Laura Gómez-Dabó, Arnau Llaurado, Daniel Sánchez-Tejerina, Victoria González, Carmen Montalvo-Olmedo, Carlos Lázaro-Hernández, Marc Rodrigo-Gisbert, Samuel López-Maza, Maider Iza-Achutegui, Lídia Giramé-Rizzo, Nuria Raguer, Raúl Juntas","doi":"10.31083/RN37744","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>We present a rare case of acute immune-mediated polyradiculoneuritis, a Guillain-Barré Syndrome (GBS) variant, manifesting as ophthalmoparesis-ataxia, facial diplegia, and acute bulbar palsy, accompanied by a unique autoimmune profile.</p><p><strong>Clinical case: </strong>A 75-year-old female developed rapidly progressive symptoms, including bilateral non-reactive mydriasis, ptosis, complete ophthalmoplegia, bilateral facial weakness, tongue immobility, palatal paralysis, limb dysmetria, ataxia, and brisk generalized tendon reflexes, all while maintaining a preserved mental state. Symptoms emerged 10 days after a probable gastrointestinal infection. Severe bulbar dysfunction necessitated orotracheal intubation and a tracheotomy. Extensive cranial nerve involvement initially suggested a brainstem lesion, with oculomotor and acute bulbar palsy as prominent signs. However, brainstem and spinal magnetic resonance imaging along with cerebrospinal fluid analysis yielded negative results. Electromyography reveled a sensorimotor demyelinating polyradiculoneuropathy, and serum testing identified IgG antibodies targeting multiple gangliosides, including the disialosyl group and terminal NeuNAc(α2-3)Gal. Treatment with intravenous immunoglobulin (IVIG) led to gradual clinical improvement.</p><p><strong>Conclusions: </strong>This case highlights a rare and severe GBS phenotype characterized by reactivity to multiple gangliosides. It highlights the role of shared ganglioside epitopes in antibody-mediated neurological damage and expands the clinical spectrum of GBS variants.</p>","PeriodicalId":21281,"journal":{"name":"Revista de neurologia","volume":"80 1","pages":"37744"},"PeriodicalIF":0.8000,"publicationDate":"2025-02-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11907704/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Revista de neurologia","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.31083/RN37744","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: We present a rare case of acute immune-mediated polyradiculoneuritis, a Guillain-Barré Syndrome (GBS) variant, manifesting as ophthalmoparesis-ataxia, facial diplegia, and acute bulbar palsy, accompanied by a unique autoimmune profile.
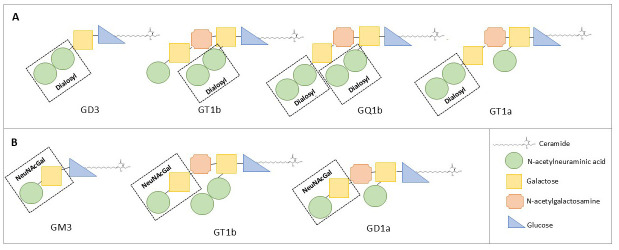
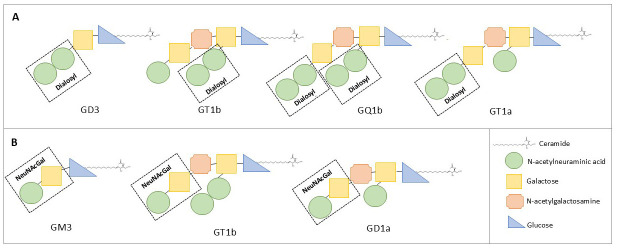
Clinical case: A 75-year-old female developed rapidly progressive symptoms, including bilateral non-reactive mydriasis, ptosis, complete ophthalmoplegia, bilateral facial weakness, tongue immobility, palatal paralysis, limb dysmetria, ataxia, and brisk generalized tendon reflexes, all while maintaining a preserved mental state. Symptoms emerged 10 days after a probable gastrointestinal infection. Severe bulbar dysfunction necessitated orotracheal intubation and a tracheotomy. Extensive cranial nerve involvement initially suggested a brainstem lesion, with oculomotor and acute bulbar palsy as prominent signs. However, brainstem and spinal magnetic resonance imaging along with cerebrospinal fluid analysis yielded negative results. Electromyography reveled a sensorimotor demyelinating polyradiculoneuropathy, and serum testing identified IgG antibodies targeting multiple gangliosides, including the disialosyl group and terminal NeuNAc(α2-3)Gal. Treatment with intravenous immunoglobulin (IVIG) led to gradual clinical improvement.
Conclusions: This case highlights a rare and severe GBS phenotype characterized by reactivity to multiple gangliosides. It highlights the role of shared ganglioside epitopes in antibody-mediated neurological damage and expands the clinical spectrum of GBS variants.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
