Does Early Diagnosis and Treatment Alter the Clinical Course of Wolman Disease? Divergent Trajectories in Two Siblings and a Consideration for Newborn Screening.
Maria Jose de Castro Lopez, Fiona J White, Victoria Holmes, Jane Roberts, Teresa H Y Wu, James A Cooper, Heather J Church, Gemma Petts, Robert F Wynn, Simon A Jones, Arunabha Ghosh
{"title":"Does Early Diagnosis and Treatment Alter the Clinical Course of Wolman Disease? Divergent Trajectories in Two Siblings and a Consideration for Newborn Screening.","authors":"Maria Jose de Castro Lopez, Fiona J White, Victoria Holmes, Jane Roberts, Teresa H Y Wu, James A Cooper, Heather J Church, Gemma Petts, Robert F Wynn, Simon A Jones, Arunabha Ghosh","doi":"10.3390/ijns11010017","DOIUrl":null,"url":null,"abstract":"<p><p>Wolman disease (WD) is a lethal disorder defined by the deficiency of the lysosomal acid lipase enzyme. Patients present with intestinal failure, malnutrition, and hepatosplenomegaly. Enzyme replacement therapy (ERT) with dietary substrate reduction (DSR) significantly improves survival. We sought to determine the outcomes of two siblings with WD treated after the onset of symptoms (sibling 1) and presymptomatic (sibling 2). A chart review was conducted on two siblings with WD treated with ERT and DSR at 4 months of age (sibling 1) and immediately after birth (sibling 2) to determine clinical outcomes based on survival, laboratory results, growth, dietary records, and gut biopsies. Sibling 1 presented with hepatosplenomegaly and liver dysfunction and developed hemophagocytic lymphohistiocytosis despite treatment. She received a bone marrow transplant at 8 months of age but died at 13 months. Sibling 2 is alive at 16 months of age with height, weight, and MUAC above the 95th centile, fully orally fed, with no gastrointestinal symptoms, normal liver function, and normal oxysterols. Sibling 2 duodenal biopsies show normal villus architecture with no foamy macrophage infiltration. Initiation of treatment prior to the onset of symptoms can prevent clinical manifestations and increase survival. The divergent trajectory in these siblings raises the question of WD's candidacy for newborn screening.</p>","PeriodicalId":14159,"journal":{"name":"International Journal of Neonatal Screening","volume":"11 1","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2025-02-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11943304/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Neonatal Screening","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ijns11010017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
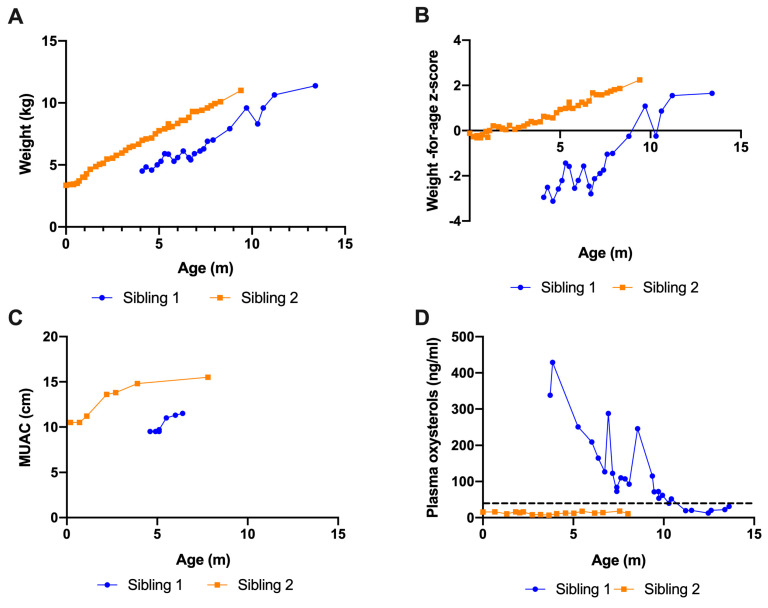
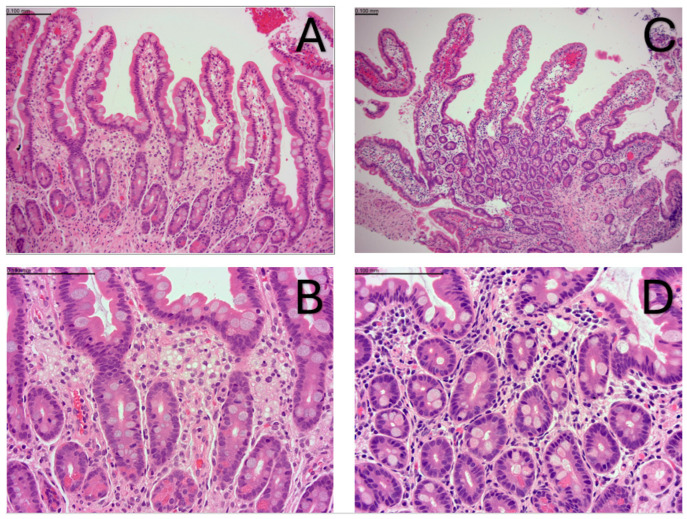
Wolman disease (WD) is a lethal disorder defined by the deficiency of the lysosomal acid lipase enzyme. Patients present with intestinal failure, malnutrition, and hepatosplenomegaly. Enzyme replacement therapy (ERT) with dietary substrate reduction (DSR) significantly improves survival. We sought to determine the outcomes of two siblings with WD treated after the onset of symptoms (sibling 1) and presymptomatic (sibling 2). A chart review was conducted on two siblings with WD treated with ERT and DSR at 4 months of age (sibling 1) and immediately after birth (sibling 2) to determine clinical outcomes based on survival, laboratory results, growth, dietary records, and gut biopsies. Sibling 1 presented with hepatosplenomegaly and liver dysfunction and developed hemophagocytic lymphohistiocytosis despite treatment. She received a bone marrow transplant at 8 months of age but died at 13 months. Sibling 2 is alive at 16 months of age with height, weight, and MUAC above the 95th centile, fully orally fed, with no gastrointestinal symptoms, normal liver function, and normal oxysterols. Sibling 2 duodenal biopsies show normal villus architecture with no foamy macrophage infiltration. Initiation of treatment prior to the onset of symptoms can prevent clinical manifestations and increase survival. The divergent trajectory in these siblings raises the question of WD's candidacy for newborn screening.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
