Gilles M Leclerc, Guy J Leclerc, Guilian Fu, Julio C Barredo
{"title":"AMPK-induced activation of Akt by AICAR is mediated by IGF-1R dependent and independent mechanisms in acute lymphoblastic leukemia.","authors":"Gilles M Leclerc, Guy J Leclerc, Guilian Fu, Julio C Barredo","doi":"10.1186/1750-2187-5-15","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Children with Acute Lymphoblastic Leukemia (ALL) diagnosed with resistant phenotypes and those who relapse have a dismal prognosis for cure. In search for novel treatment strategies, we identified the AMP activated protein kinase (AMPK) as a potential drug target based on its effects on cell growth and survival. We have shown previously that AICAR-induced AMPK activation also induced a compensatory survival mechanism via PI3K/Akt signaling.</p><p><strong>Results: </strong>In the present study, we further investigated the downstream signaling induced by AMPK activation in ALL cells. We found that AICAR-induced AMPK activation resulted in up-regulation of P-Akt (Ser473 and Thr308) and decrease of P-mTOR (Ser2448) expression and downstream signaling. We determined that activation of P-Akt (Thr308) was mediated by AMPK-induced IGF-1R activation via phosphorylation of the insulin receptor substrate-1 (IRS-1) at Ser794. Inhibition of IGF-1R signaling using the tyrosine kinase inhibitor HNMPA(AM)3 resulted in significant decrease in P-IRS-1 (Ser794) and P-Akt (Thr308). Co-treatment of AICAR plus HNMPA(AM)3 prevented AMPK-induced up-regulation of P-Akt (Thr308) but did not alter the activation of P-Akt (Ser473). Inhibition of AMPK using compound-C resulted in decreased P-Akt expression at both residues, suggesting a central role for AMPK in Akt activation. In addition, inhibition of IGF-1R signaling in ALL cells resulted in cell growth arrest and apoptosis. Additional Western blots revealed that P-IGF-1R (Tyr1131) and P-IRS-1 (Ser794) levels were higher in NALM6 (Bp-ALL) than CEM (T-ALL), and found differences in IGF-1R signaling within Bp-ALL cell line models NALM6, REH (TEL-AML1, [t(12;21)]), and SupB15 (BCR-ABL, [t(9;22)]). In these models, higher sensitivity to IGF-1R inhibitors correlated with increased levels of IGF-1R expression. Combined therapy simultaneously targeting IGF-1R, AMPK, Akt, and mTOR pathways resulted in synergistic growth inhibition and cell death.</p><p><strong>Conclusions: </strong>Our study demonstrates that AMPK activates Akt through IGF-1R dependent and independent mechanisms. Co-targeting IGF-1R and related downstream metabolic and oncogenic signaling pathways represent a potential strategy for future translation into novel ALL therapies.</p>","PeriodicalId":35051,"journal":{"name":"Journal of Molecular Signaling","volume":"5 ","pages":"15"},"PeriodicalIF":0.0000,"publicationDate":"2010-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1750-2187-5-15","citationCount":"85","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Signaling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1750-2187-5-15","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 85
Abstract
Background: Children with Acute Lymphoblastic Leukemia (ALL) diagnosed with resistant phenotypes and those who relapse have a dismal prognosis for cure. In search for novel treatment strategies, we identified the AMP activated protein kinase (AMPK) as a potential drug target based on its effects on cell growth and survival. We have shown previously that AICAR-induced AMPK activation also induced a compensatory survival mechanism via PI3K/Akt signaling.
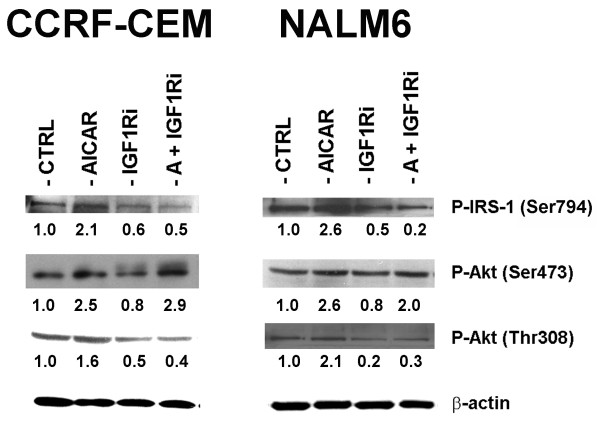
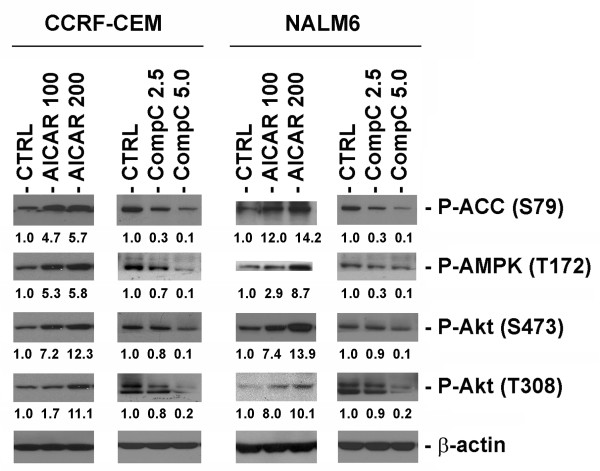
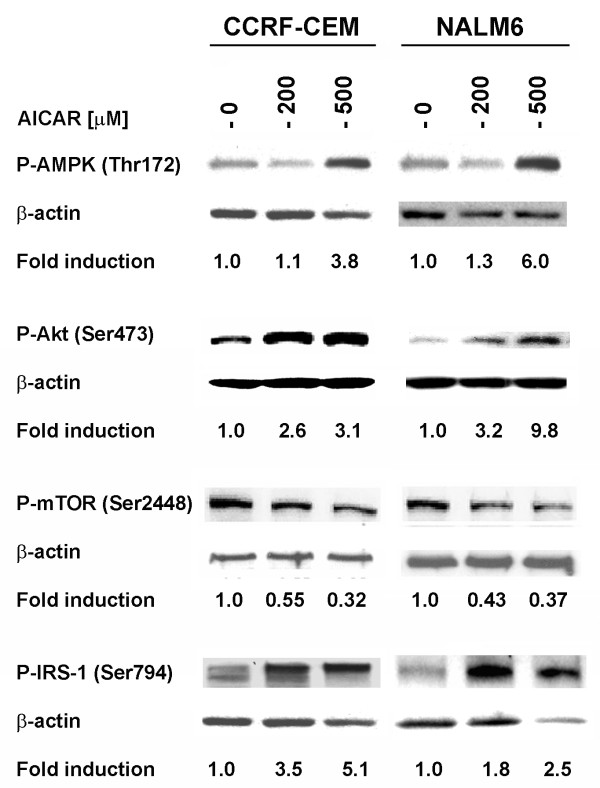
Results: In the present study, we further investigated the downstream signaling induced by AMPK activation in ALL cells. We found that AICAR-induced AMPK activation resulted in up-regulation of P-Akt (Ser473 and Thr308) and decrease of P-mTOR (Ser2448) expression and downstream signaling. We determined that activation of P-Akt (Thr308) was mediated by AMPK-induced IGF-1R activation via phosphorylation of the insulin receptor substrate-1 (IRS-1) at Ser794. Inhibition of IGF-1R signaling using the tyrosine kinase inhibitor HNMPA(AM)3 resulted in significant decrease in P-IRS-1 (Ser794) and P-Akt (Thr308). Co-treatment of AICAR plus HNMPA(AM)3 prevented AMPK-induced up-regulation of P-Akt (Thr308) but did not alter the activation of P-Akt (Ser473). Inhibition of AMPK using compound-C resulted in decreased P-Akt expression at both residues, suggesting a central role for AMPK in Akt activation. In addition, inhibition of IGF-1R signaling in ALL cells resulted in cell growth arrest and apoptosis. Additional Western blots revealed that P-IGF-1R (Tyr1131) and P-IRS-1 (Ser794) levels were higher in NALM6 (Bp-ALL) than CEM (T-ALL), and found differences in IGF-1R signaling within Bp-ALL cell line models NALM6, REH (TEL-AML1, [t(12;21)]), and SupB15 (BCR-ABL, [t(9;22)]). In these models, higher sensitivity to IGF-1R inhibitors correlated with increased levels of IGF-1R expression. Combined therapy simultaneously targeting IGF-1R, AMPK, Akt, and mTOR pathways resulted in synergistic growth inhibition and cell death.
Conclusions: Our study demonstrates that AMPK activates Akt through IGF-1R dependent and independent mechanisms. Co-targeting IGF-1R and related downstream metabolic and oncogenic signaling pathways represent a potential strategy for future translation into novel ALL therapies.
期刊介绍:
Journal of Molecular Signaling is an open access, peer-reviewed online journal that encompasses all aspects of molecular signaling. Molecular signaling is an exponentially growing field that encompasses different molecular aspects of cell signaling underlying normal and pathological conditions. Specifically, the research area of the journal is on the normal or aberrant molecular mechanisms involving receptors, G-proteins, kinases, phosphatases, and transcription factors in regulating cell proliferation, differentiation, apoptosis, and oncogenesis in mammalian cells. This area also covers the genetic and epigenetic changes that modulate the signaling properties of cells and the resultant physiological conditions.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
