Joseph F Schad, Kate R Meltzer, Michael R Hicks, David S Beutler, Thanh V Cao, Paul R Standley
{"title":"Cyclic strain upregulates VEGF and attenuates proliferation of vascular smooth muscle cells.","authors":"Joseph F Schad, Kate R Meltzer, Michael R Hicks, David S Beutler, Thanh V Cao, Paul R Standley","doi":"10.1186/2045-824X-3-21","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Vascular smooth muscle cell (VSMC) hypertrophy and proliferation occur in response to strain-induced local and systemic inflammatory cytokines and growth factors which may contribute to hypertension, atherosclerosis, and restenosis. We hypothesize VSMC strain, modeling normotensive arterial pressure waveforms in vitro, results in attenuated proliferative and increased hypertrophic responses 48 hrs post-strain.</p><p><strong>Methods: </strong>Using Flexcell Bioflex Systems we determined the morphological, hyperplastic and hypertrophic responses of non-strained and biomechanically strained cultured rat A7R5 VSMC. We measured secretion of nitric oxide, key cytokine/growth factors and intracellular mediators involved in VSMC proliferation via fluorescence spectroscopy and protein microarrays. We also investigated the potential roles of VEGF on VSMC strain-induced proliferation.</p><p><strong>Results: </strong>Protein microarrays revealed significant increases in VEGF secretion in response to 18 hours mechanical strain, a result that ELISA data corroborated. Apoptosis-inducing nitric oxide (NO) levels also increased 43% 48 hrs post-strain. Non-strained cells incubated with exogenous VEGF did not reproduce the antimitogenic effect. However, anti-VEGF reversed the antimitogenic effect of mechanical strain. Antibody microarrays of strained VSMC lysates revealed MEK1, MEK2, phospo-MEK1T385, T291, T298, phospho-Erk1/2T202+Y204/T185+T187, and PKC isoforms expression were universally increased, suggesting a proliferative/inflammatory signaling state. Conversely, VSMC strain decreased expression levels of Cdk1, Cdk2, Cdk4, and Cdk6 by 25-50% suggesting a partially inhibited proliferative signaling cascade.</p><p><strong>Conclusions: </strong>Subjecting VSMC to cyclic biomechanical strain in vitro promotes cell hypertrophy while attenuating cellular proliferation. We also report an upregulation of MEK and ERK activation suggestive of a proliferative phenotype. Hhowever, the proliferative response appears to be aborogated by enhanced antimitogenic cytokine VEGF, NO secretion and downregulation of Cdk expression. Although exogenous VEGF alone is not sufficient to promote the quiescent VSMC phenotype, we provide evidence suggesting that strain is a necessary component to induce VSMC response to the antimitogenic effects of VEGF. Taken together these data indicate that VEGF plays a critical role in mechanical strain-induced VSMC proliferation and vessel wall remodeling. Whether VEGF and/or NO inhibit signaling distal to Erk 1/2 is currently under investigation.</p>","PeriodicalId":23948,"journal":{"name":"Vascular Cell","volume":"3 ","pages":"21"},"PeriodicalIF":0.0000,"publicationDate":"2011-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2045-824X-3-21","citationCount":"35","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Vascular Cell","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2045-824X-3-21","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Neuroscience","Score":null,"Total":0}
引用次数: 35
Abstract
Objective: Vascular smooth muscle cell (VSMC) hypertrophy and proliferation occur in response to strain-induced local and systemic inflammatory cytokines and growth factors which may contribute to hypertension, atherosclerosis, and restenosis. We hypothesize VSMC strain, modeling normotensive arterial pressure waveforms in vitro, results in attenuated proliferative and increased hypertrophic responses 48 hrs post-strain.
Methods: Using Flexcell Bioflex Systems we determined the morphological, hyperplastic and hypertrophic responses of non-strained and biomechanically strained cultured rat A7R5 VSMC. We measured secretion of nitric oxide, key cytokine/growth factors and intracellular mediators involved in VSMC proliferation via fluorescence spectroscopy and protein microarrays. We also investigated the potential roles of VEGF on VSMC strain-induced proliferation.
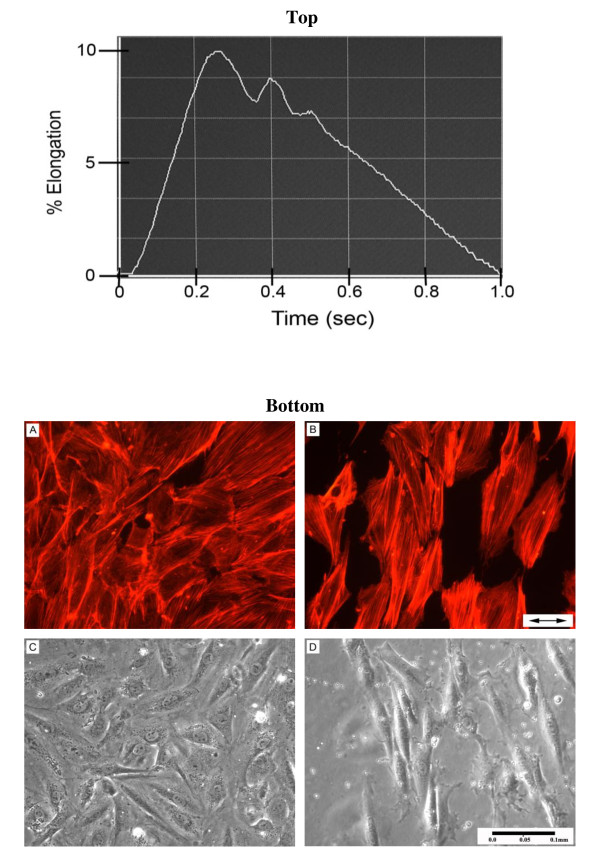
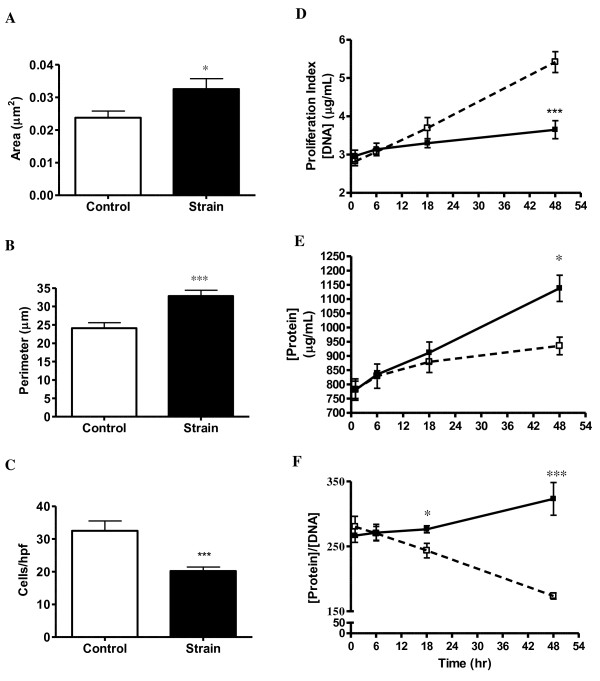
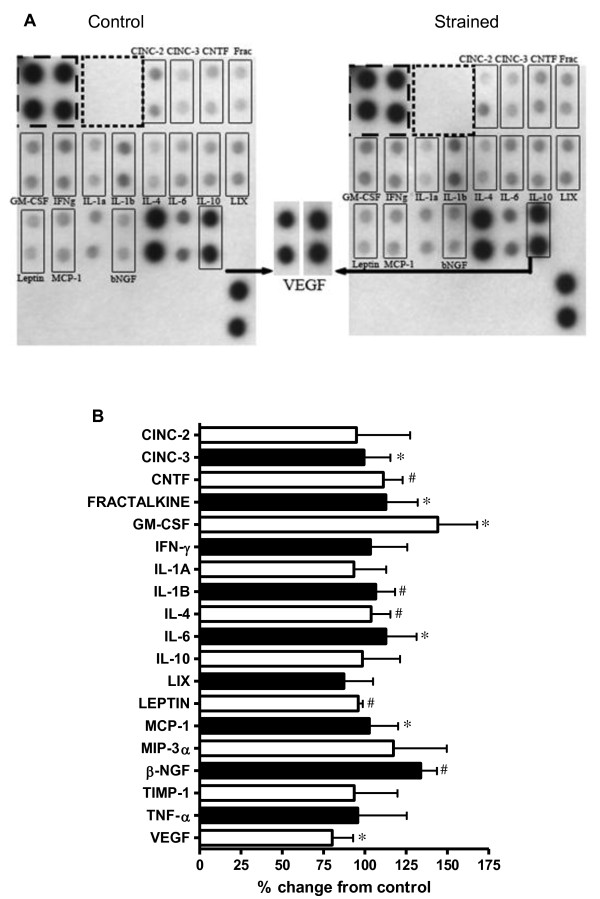
Results: Protein microarrays revealed significant increases in VEGF secretion in response to 18 hours mechanical strain, a result that ELISA data corroborated. Apoptosis-inducing nitric oxide (NO) levels also increased 43% 48 hrs post-strain. Non-strained cells incubated with exogenous VEGF did not reproduce the antimitogenic effect. However, anti-VEGF reversed the antimitogenic effect of mechanical strain. Antibody microarrays of strained VSMC lysates revealed MEK1, MEK2, phospo-MEK1T385, T291, T298, phospho-Erk1/2T202+Y204/T185+T187, and PKC isoforms expression were universally increased, suggesting a proliferative/inflammatory signaling state. Conversely, VSMC strain decreased expression levels of Cdk1, Cdk2, Cdk4, and Cdk6 by 25-50% suggesting a partially inhibited proliferative signaling cascade.
Conclusions: Subjecting VSMC to cyclic biomechanical strain in vitro promotes cell hypertrophy while attenuating cellular proliferation. We also report an upregulation of MEK and ERK activation suggestive of a proliferative phenotype. Hhowever, the proliferative response appears to be aborogated by enhanced antimitogenic cytokine VEGF, NO secretion and downregulation of Cdk expression. Although exogenous VEGF alone is not sufficient to promote the quiescent VSMC phenotype, we provide evidence suggesting that strain is a necessary component to induce VSMC response to the antimitogenic effects of VEGF. Taken together these data indicate that VEGF plays a critical role in mechanical strain-induced VSMC proliferation and vessel wall remodeling. Whether VEGF and/or NO inhibit signaling distal to Erk 1/2 is currently under investigation.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
