{"title":"Protein-x of hepatitis B virus in interaction with CCAAT/enhancer-binding protein α (C/EBPα)--an in silico analysis approach.","authors":"Ashraf Mohamadkhani, Parisa Shahnazari, Zarrin Minuchehr, Armin Madadkar-Sobhani, Mahmoud Jeddi Tehrani, Ferdous Rastgar Jazii, Hossein Poustchi","doi":"10.1186/1742-4682-8-41","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Even though many functions of protein-x from the Hepatitis B virus (HBV) have been revealed, the nature of protein-x is yet unknown. This protein is well-known for its transactivation activity through interaction with several cellular transcription factors, it is also known as an oncogene. In this work, we have presented computational approaches to design a model to show the structure of protein-x and its respective binding sites associated with the CCAAT/enhancer-binding protein α (C/EBPα). C/EBPα belongs to the bZip family of transcription factors, which activates transcription of several genes through its binding sites in liver and fat cells. The C/EBPα has been shown to bind and modulate enhancer I and the enhancer II/core promoter of HBV. In this study using the bioinformatics tools we tried to present a reliable model for the protein-x interaction with C/EBPα.</p><p><strong>Results: </strong>The amino acid sequence of protein-x was extracted from UniProt [UniProt:Q80IU5] and the x-ray crystal structure of the partial CCAAT-enhancer α [PDB:1NWQ] was retrieved from the Protein Data Bank (PDB). Similarity search for protein-x was carried out by psi-blast and bl2seq using NCBI [GenBank: BAC65106.1] and Local Meta-Threading-Server (LOMETS) was used as a threading server for determining the maximum tertiary structure similarities. Advanced MODELLER was implemented to design a comparative model, however, due to the lack of a suitable template, Quark was used for ab initio tertiary structure prediction.The PDB-blast search indicated a maximum of 23% sequence identity and 33% similarity with crystal structure of the porcine reproductive and respiratory syndrome virus leader protease Nsp1α [PDB:3IFU]. This meant that protein-x does not have a suitable template to predict its tertiary structure using comparative modeling tools, therefore we used QUARK as an ab initio 3D prediction approach. Docking results from the ab initio tertiary structure of protein-x and crystal structure of the C/EBPα- DNA region [PDB:1NWQ] illustrated the protein-binding site interactions. Indeed, the N-terminal part of 1NWQ has a high affinity for certain regions in protein-x (e.g. from Ala76 to Ser101 and Thr105 to Glu125).</p><p><strong>Conclusion: </strong>In this study, we predicted the structure of protein-x of HBV in interaction with C/EBPα. The docking results showed that protein-x has an interaction synergy with C/EBPα. However, despite previous experimental data, protein-x was found to interact with DNA. This can lead to a better understanding of the function of protein-x and may provide an opportunity to use it as a therapeutic target.</p>","PeriodicalId":51195,"journal":{"name":"Theoretical Biology and Medical Modelling","volume":" ","pages":"41"},"PeriodicalIF":0.0000,"publicationDate":"2011-10-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1742-4682-8-41","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Biology and Medical Modelling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1742-4682-8-41","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Mathematics","Score":null,"Total":0}
引用次数: 4
Abstract
Background: Even though many functions of protein-x from the Hepatitis B virus (HBV) have been revealed, the nature of protein-x is yet unknown. This protein is well-known for its transactivation activity through interaction with several cellular transcription factors, it is also known as an oncogene. In this work, we have presented computational approaches to design a model to show the structure of protein-x and its respective binding sites associated with the CCAAT/enhancer-binding protein α (C/EBPα). C/EBPα belongs to the bZip family of transcription factors, which activates transcription of several genes through its binding sites in liver and fat cells. The C/EBPα has been shown to bind and modulate enhancer I and the enhancer II/core promoter of HBV. In this study using the bioinformatics tools we tried to present a reliable model for the protein-x interaction with C/EBPα.
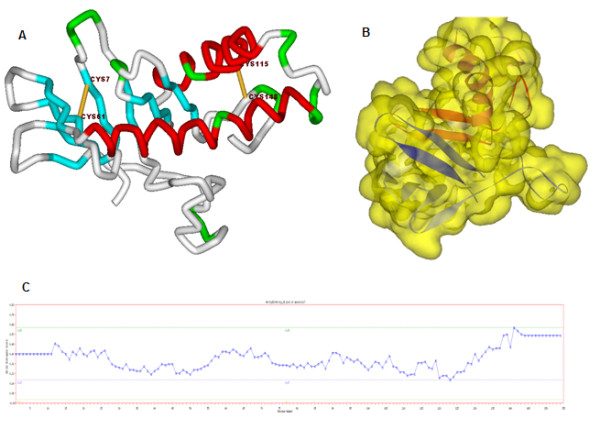
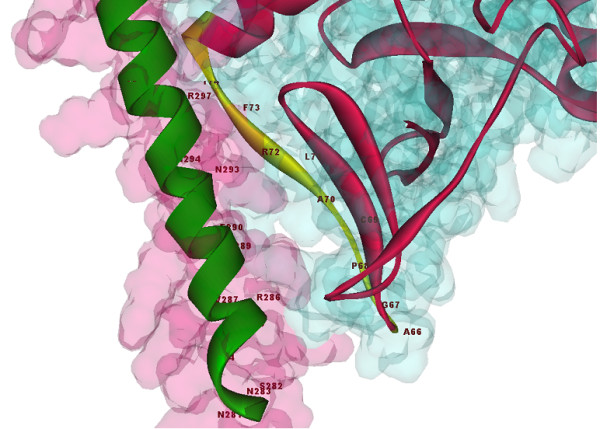
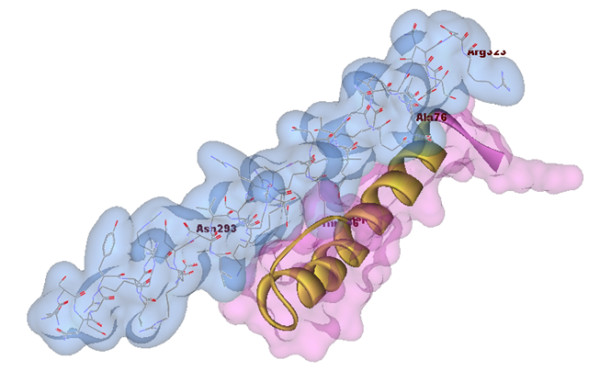
Results: The amino acid sequence of protein-x was extracted from UniProt [UniProt:Q80IU5] and the x-ray crystal structure of the partial CCAAT-enhancer α [PDB:1NWQ] was retrieved from the Protein Data Bank (PDB). Similarity search for protein-x was carried out by psi-blast and bl2seq using NCBI [GenBank: BAC65106.1] and Local Meta-Threading-Server (LOMETS) was used as a threading server for determining the maximum tertiary structure similarities. Advanced MODELLER was implemented to design a comparative model, however, due to the lack of a suitable template, Quark was used for ab initio tertiary structure prediction.The PDB-blast search indicated a maximum of 23% sequence identity and 33% similarity with crystal structure of the porcine reproductive and respiratory syndrome virus leader protease Nsp1α [PDB:3IFU]. This meant that protein-x does not have a suitable template to predict its tertiary structure using comparative modeling tools, therefore we used QUARK as an ab initio 3D prediction approach. Docking results from the ab initio tertiary structure of protein-x and crystal structure of the C/EBPα- DNA region [PDB:1NWQ] illustrated the protein-binding site interactions. Indeed, the N-terminal part of 1NWQ has a high affinity for certain regions in protein-x (e.g. from Ala76 to Ser101 and Thr105 to Glu125).
Conclusion: In this study, we predicted the structure of protein-x of HBV in interaction with C/EBPα. The docking results showed that protein-x has an interaction synergy with C/EBPα. However, despite previous experimental data, protein-x was found to interact with DNA. This can lead to a better understanding of the function of protein-x and may provide an opportunity to use it as a therapeutic target.
期刊介绍:
Theoretical Biology and Medical Modelling is an open access peer-reviewed journal adopting a broad definition of "biology" and focusing on theoretical ideas and models associated with developments in biology and medicine. Mathematicians, biologists and clinicians of various specialisms, philosophers and historians of science are all contributing to the emergence of novel concepts in an age of systems biology, bioinformatics and computer modelling. This is the field in which Theoretical Biology and Medical Modelling operates. We welcome submissions that are technically sound and offering either improved understanding in biology and medicine or progress in theory or method.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
