Qiu-Lan Ma, Fusheng Yang, Sally A Frautschy, Greg M Cole
{"title":"PAK in Alzheimer disease, Huntington disease and X-linked mental retardation.","authors":"Qiu-Lan Ma, Fusheng Yang, Sally A Frautschy, Greg M Cole","doi":"10.4161/cl.21602","DOIUrl":null,"url":null,"abstract":"<p><p>Developmental cognitive deficits including X-linked mental retardation (XLMR) can be caused by mutations in P21-activated kinase 3 (PAK3) that disrupt actin dynamics in dendritic spines. Neurodegenerative diseases such as Alzheimer disease (AD), where both PAK1 and PAK3 are dysregulated, may share final common pathways with XLMR. Independent of familial mutation, cognitive deficits emerging with aging, notably AD, begin after decades of normal function. This prolonged prodromal period involves the buildup of amyloid-β (Aβ) extracellular plaques and intraneuronal neurofibrillary tangles (NFT). Subsequently region dependent deficits in synapses, dendritic spines and cognition coincide with dysregulation in PAK1 and PAK. Specifically proximal to decline, cytoplasmic levels of actin-regulating Rho GTPase and PAK1 kinase are decreased in moderate to severe AD, while aberrant activation and translocation of PAK1 appears around the onset of cognitive deficits. Downstream to PAK1, LIM kinase inactivates cofilin, contributing to cofilin pathology, while the activation of Rho-dependent kinase ROCK increases Aβ production. Aβ activation of fyn disrupts neuronal PAK1 and ROCK-mediated signaling, resulting in synaptic deficits. Reductions in PAK1 by the anti-amyloid compound curcumin suppress synaptotoxicity. Similarly other neurological disorders, including Huntington disease (HD) show dysregulation of PAKs. PAK1 modulates mutant huntingtin toxicity by enhancing huntingtin aggregation, and inhibition of PAK activity protects HD as well as fragile X syndrome (FXS) symptoms. Since PAK plays critical roles in learning and memory and is disrupted in many cognitive disorders, targeting PAK signaling in AD, HD and XLMR may be a novel common therapeutic target for AD, HD and XLMR.</p>","PeriodicalId":72547,"journal":{"name":"Cellular logistics","volume":"2 2","pages":"117-125"},"PeriodicalIF":0.0000,"publicationDate":"2012-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4161/cl.21602","citationCount":"72","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular logistics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4161/cl.21602","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 72
Abstract
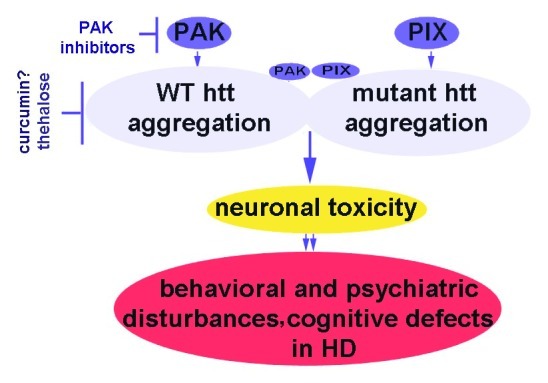
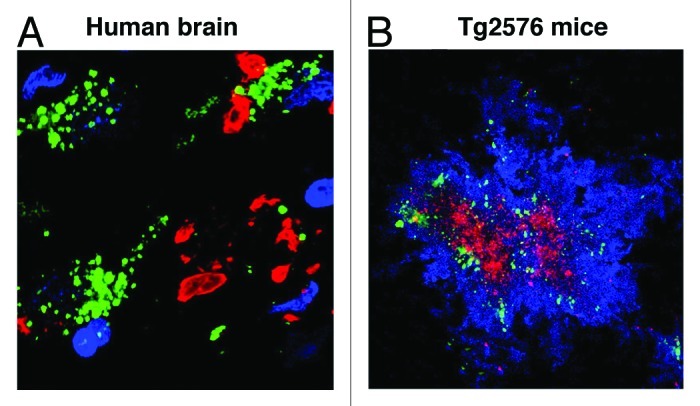
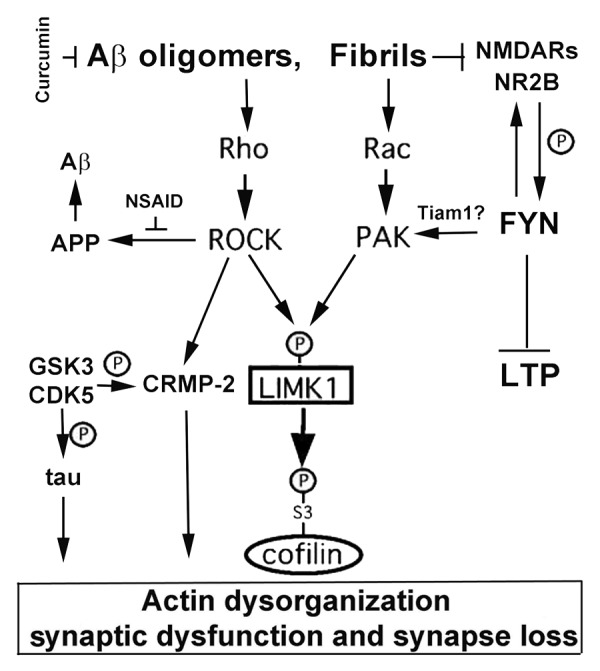
Developmental cognitive deficits including X-linked mental retardation (XLMR) can be caused by mutations in P21-activated kinase 3 (PAK3) that disrupt actin dynamics in dendritic spines. Neurodegenerative diseases such as Alzheimer disease (AD), where both PAK1 and PAK3 are dysregulated, may share final common pathways with XLMR. Independent of familial mutation, cognitive deficits emerging with aging, notably AD, begin after decades of normal function. This prolonged prodromal period involves the buildup of amyloid-β (Aβ) extracellular plaques and intraneuronal neurofibrillary tangles (NFT). Subsequently region dependent deficits in synapses, dendritic spines and cognition coincide with dysregulation in PAK1 and PAK. Specifically proximal to decline, cytoplasmic levels of actin-regulating Rho GTPase and PAK1 kinase are decreased in moderate to severe AD, while aberrant activation and translocation of PAK1 appears around the onset of cognitive deficits. Downstream to PAK1, LIM kinase inactivates cofilin, contributing to cofilin pathology, while the activation of Rho-dependent kinase ROCK increases Aβ production. Aβ activation of fyn disrupts neuronal PAK1 and ROCK-mediated signaling, resulting in synaptic deficits. Reductions in PAK1 by the anti-amyloid compound curcumin suppress synaptotoxicity. Similarly other neurological disorders, including Huntington disease (HD) show dysregulation of PAKs. PAK1 modulates mutant huntingtin toxicity by enhancing huntingtin aggregation, and inhibition of PAK activity protects HD as well as fragile X syndrome (FXS) symptoms. Since PAK plays critical roles in learning and memory and is disrupted in many cognitive disorders, targeting PAK signaling in AD, HD and XLMR may be a novel common therapeutic target for AD, HD and XLMR.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
