Brian Moldover, Ada Solidar, Christa Montgomery, Henry Miziorko, Jeff Murphy, Gerald J Wyckoff
{"title":"ChemVassa: A New Method for Identifying Small Molecule Hits in Drug Discovery.","authors":"Brian Moldover, Ada Solidar, Christa Montgomery, Henry Miziorko, Jeff Murphy, Gerald J Wyckoff","doi":"10.2174/1874104501206010029","DOIUrl":null,"url":null,"abstract":"<p><p>ChemVassa, a new chemical structure search technology, was developed to allow rapid in silico screening of compounds for hit and hit-to-lead identification in drug development. It functions by using a novel type of molecular descriptor that examines, in part, the structure of the small molecule undergoing analysis, yielding its \"information signature.\" This descriptor takes into account the atoms, bonds, and their positions in 3-dimensional space. For the present study, a database of ChemVassa molecular descriptors was generated for nearly 16 million compounds (from the ZINC database and other compound sources), then an algorithm was developed that allows rapid similarity searching of the database using a query molecular descriptor (e.g., the signature of atorvastatin, below). A scoring metric then allowed ranking of the search results. We used these tools to search a subset of drug-like molecules using the signature of a commercially successful statin, atorvastatin (Lipitor™). The search identified ten novel compounds, two of which have been demonstrated to interact with HMG-CoA reductase, the macromolecular target of atorvastatin. In particular, one compound discussed in the results section tested successfully with an IC50 of less than 100uM and a completely novel structure relative to known inhibitors. Interactions were validated using computational molecular docking and an Hmg-CoA reductase activity assay. The rapidity and low cost of the methodology, and the novel structure of the interactors, suggests this is a highly favorable new method for hit generation.</p>","PeriodicalId":39133,"journal":{"name":"Open Medicinal Chemistry Journal","volume":"6 ","pages":"29-34"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/f8/c7/TOMCJ-6-29.PMC3601345.pdf","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Open Medicinal Chemistry Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2174/1874104501206010029","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2012/11/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Pharmacology, Toxicology and Pharmaceutics","Score":null,"Total":0}
引用次数: 4
Abstract
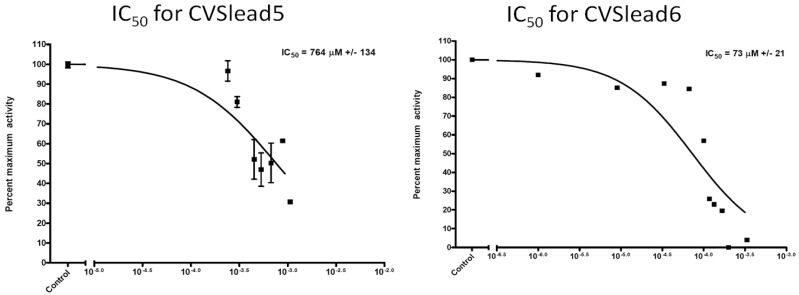
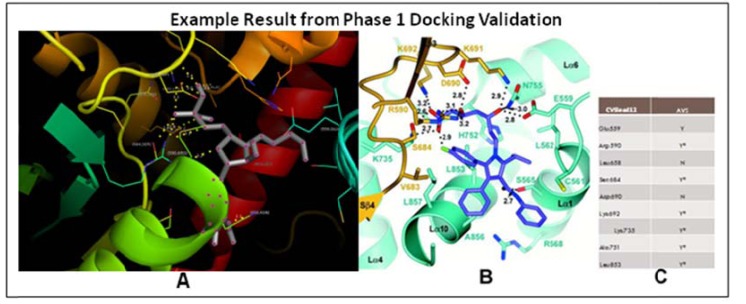
ChemVassa, a new chemical structure search technology, was developed to allow rapid in silico screening of compounds for hit and hit-to-lead identification in drug development. It functions by using a novel type of molecular descriptor that examines, in part, the structure of the small molecule undergoing analysis, yielding its "information signature." This descriptor takes into account the atoms, bonds, and their positions in 3-dimensional space. For the present study, a database of ChemVassa molecular descriptors was generated for nearly 16 million compounds (from the ZINC database and other compound sources), then an algorithm was developed that allows rapid similarity searching of the database using a query molecular descriptor (e.g., the signature of atorvastatin, below). A scoring metric then allowed ranking of the search results. We used these tools to search a subset of drug-like molecules using the signature of a commercially successful statin, atorvastatin (Lipitor™). The search identified ten novel compounds, two of which have been demonstrated to interact with HMG-CoA reductase, the macromolecular target of atorvastatin. In particular, one compound discussed in the results section tested successfully with an IC50 of less than 100uM and a completely novel structure relative to known inhibitors. Interactions were validated using computational molecular docking and an Hmg-CoA reductase activity assay. The rapidity and low cost of the methodology, and the novel structure of the interactors, suggests this is a highly favorable new method for hit generation.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
