Fabian Schmich, Ewa Szczurek, Saskia Kreibich, Sabrina Dilling, Daniel Andritschke, Alain Casanova, Shyan Huey Low, Simone Eicher, Simone Muntwiler, Mario Emmenlauer, Pauli Rämö, Raquel Conde-Alvarez, Christian von Mering, Wolf-Dietrich Hardt, Christoph Dehio, Niko Beerenwinkel
{"title":"gespeR: a statistical model for deconvoluting off-target-confounded RNA interference screens.","authors":"Fabian Schmich, Ewa Szczurek, Saskia Kreibich, Sabrina Dilling, Daniel Andritschke, Alain Casanova, Shyan Huey Low, Simone Eicher, Simone Muntwiler, Mario Emmenlauer, Pauli Rämö, Raquel Conde-Alvarez, Christian von Mering, Wolf-Dietrich Hardt, Christoph Dehio, Niko Beerenwinkel","doi":"10.1186/s13059-015-0783-1","DOIUrl":null,"url":null,"abstract":"<p><p>Small interfering RNAs (siRNAs) exhibit strong off-target effects, which confound the gene-level interpretation of RNA interference screens and thus limit their utility for functional genomics studies. Here, we present gespeR, a statistical model for reconstructing individual, gene-specific phenotypes. Using 115,878 siRNAs, single and pooled, from three companies in three pathogen infection screens, we demonstrate that deconvolution of image-based phenotypes substantially improves the reproducibility between independent siRNA sets targeting the same genes. Genes selected and prioritized by gespeR are validated and shown to constitute biologically relevant components of pathogen entry mechanisms and TGF-β signaling. gespeR is available as a Bioconductor R-package. </p>","PeriodicalId":48922,"journal":{"name":"Genome Biology","volume":"16 ","pages":"220"},"PeriodicalIF":12.3000,"publicationDate":"2015-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13059-015-0783-1","citationCount":"34","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13059-015-0783-1","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 34
Abstract
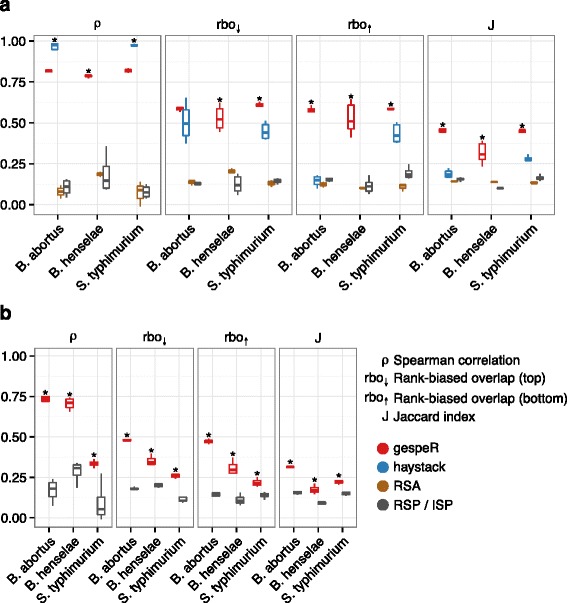
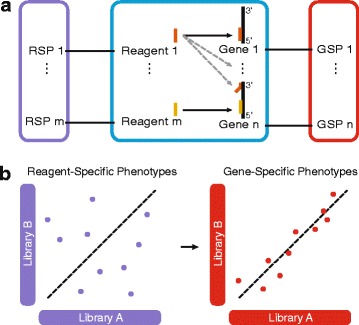
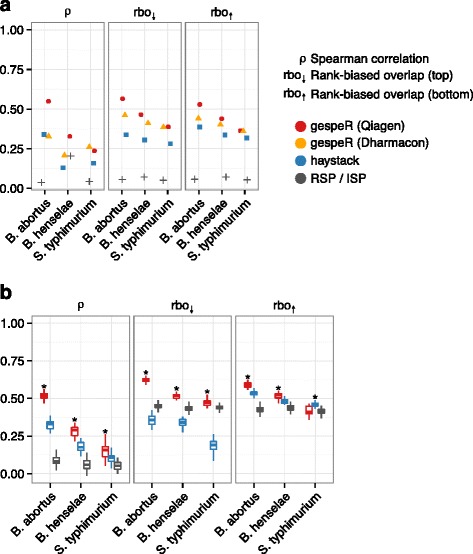
Small interfering RNAs (siRNAs) exhibit strong off-target effects, which confound the gene-level interpretation of RNA interference screens and thus limit their utility for functional genomics studies. Here, we present gespeR, a statistical model for reconstructing individual, gene-specific phenotypes. Using 115,878 siRNAs, single and pooled, from three companies in three pathogen infection screens, we demonstrate that deconvolution of image-based phenotypes substantially improves the reproducibility between independent siRNA sets targeting the same genes. Genes selected and prioritized by gespeR are validated and shown to constitute biologically relevant components of pathogen entry mechanisms and TGF-β signaling. gespeR is available as a Bioconductor R-package.
期刊介绍:
Genome Biology is a leading research journal that focuses on the study of biology and biomedicine from a genomic and post-genomic standpoint. The journal consistently publishes outstanding research across various areas within these fields.
With an impressive impact factor of 12.3 (2022), Genome Biology has earned its place as the 3rd highest-ranked research journal in the Genetics and Heredity category, according to Thomson Reuters. Additionally, it is ranked 2nd among research journals in the Biotechnology and Applied Microbiology category. It is important to note that Genome Biology is the top-ranking open access journal in this category.
In summary, Genome Biology sets a high standard for scientific publications in the field, showcasing cutting-edge research and earning recognition among its peers.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
