Shauna A Rasmussen, Michelle Kwon, Jeffrey D Pressly, Joe B Blumer, Kevin R Regner, Frank Park
{"title":"Activator of G-protein Signaling 3 Controls Renal Epithelial Cell Survival and ERK5 Activation.","authors":"Shauna A Rasmussen, Michelle Kwon, Jeffrey D Pressly, Joe B Blumer, Kevin R Regner, Frank Park","doi":"10.5334/1750-2187-10-5","DOIUrl":null,"url":null,"abstract":"<p><p>Activator of G-protein signaling 3 (AGS3) is an accessory protein that functions to regulate the activation status of heterotrimeric G-protein subunits. To date, however, the downstream signaling pathways regulated by AGS3 remain to be fully elucidated, particularly in renal epithelial cells. In the present study, normal rat kidney (NRK-52E) proximal tubular epithelial cells were genetically modified to regulate the expression of AGS3 to investigate its role on MAPK and mTOR signaling to control epithelial cell number. Knockdown of endogenous AGS3 protein was associated with a reduced phosphorylated form of ERK5 and increased apoptosis as determined by elevated cleaved caspase-3. In the presence of the ERK5 inhibitor, BIX02189, a significant 2-fold change (P < 0.05) in G2/M transition state was detected compared to control conditions. Neither of the other MAPK, ERK1/2 or p38 MAPK, nor another pro-survival pathway, mTOR, was significantly altered by the changes in AGS3 protein levels in the renal epithelial cells. The selective ERK5 inhibitor, BIX02189, was found to dose-dependently reduce NRK cell number by up to 41% (P < 0.05) compared to control cells. In summary, these findings demonstrated that cell viability was regulated by AGS3 and was associated with ERK5 activation in renal epithelial cells. </p>","PeriodicalId":35051,"journal":{"name":"Journal of Molecular Signaling","volume":" ","pages":"6"},"PeriodicalIF":0.0000,"publicationDate":"2015-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4831271/pdf/","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Signaling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5334/1750-2187-10-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 4
Abstract
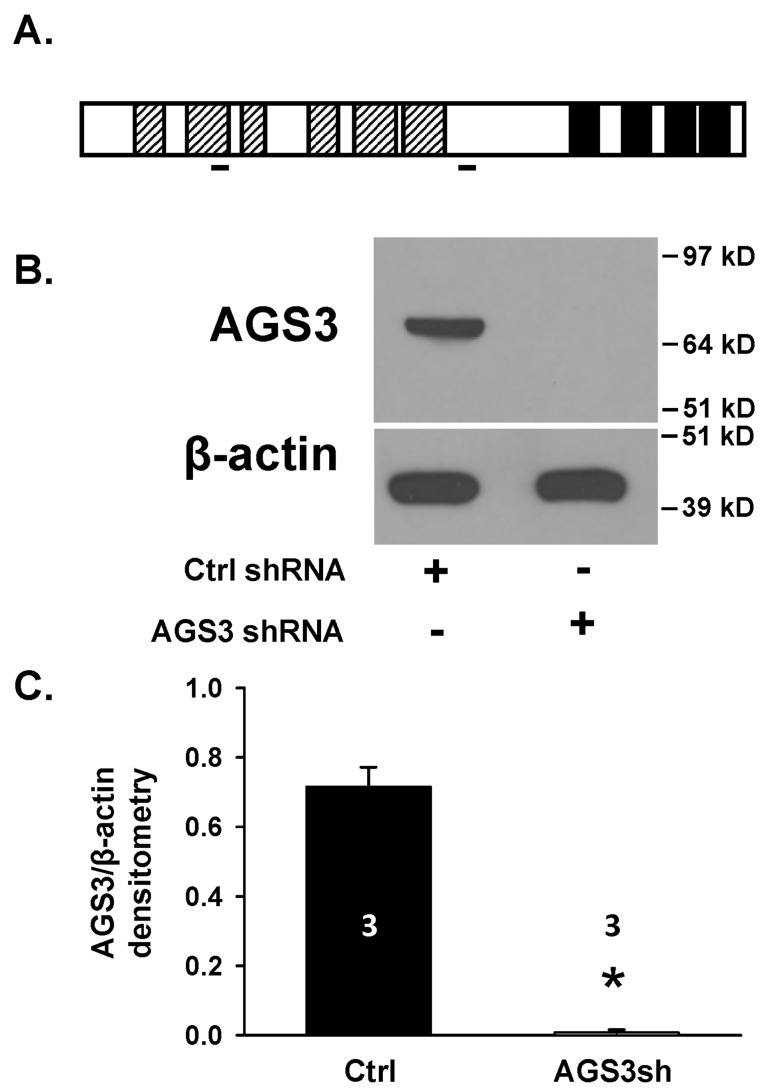
Activator of G-protein signaling 3 (AGS3) is an accessory protein that functions to regulate the activation status of heterotrimeric G-protein subunits. To date, however, the downstream signaling pathways regulated by AGS3 remain to be fully elucidated, particularly in renal epithelial cells. In the present study, normal rat kidney (NRK-52E) proximal tubular epithelial cells were genetically modified to regulate the expression of AGS3 to investigate its role on MAPK and mTOR signaling to control epithelial cell number. Knockdown of endogenous AGS3 protein was associated with a reduced phosphorylated form of ERK5 and increased apoptosis as determined by elevated cleaved caspase-3. In the presence of the ERK5 inhibitor, BIX02189, a significant 2-fold change (P < 0.05) in G2/M transition state was detected compared to control conditions. Neither of the other MAPK, ERK1/2 or p38 MAPK, nor another pro-survival pathway, mTOR, was significantly altered by the changes in AGS3 protein levels in the renal epithelial cells. The selective ERK5 inhibitor, BIX02189, was found to dose-dependently reduce NRK cell number by up to 41% (P < 0.05) compared to control cells. In summary, these findings demonstrated that cell viability was regulated by AGS3 and was associated with ERK5 activation in renal epithelial cells.
期刊介绍:
Journal of Molecular Signaling is an open access, peer-reviewed online journal that encompasses all aspects of molecular signaling. Molecular signaling is an exponentially growing field that encompasses different molecular aspects of cell signaling underlying normal and pathological conditions. Specifically, the research area of the journal is on the normal or aberrant molecular mechanisms involving receptors, G-proteins, kinases, phosphatases, and transcription factors in regulating cell proliferation, differentiation, apoptosis, and oncogenesis in mammalian cells. This area also covers the genetic and epigenetic changes that modulate the signaling properties of cells and the resultant physiological conditions.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
