Nathan D. Olson , Steven P. Lund , Justin M. Zook , Fabiola Rojas-Cornejo , Brian Beck , Carole Foy , Jim Huggett , Alexandra S. Whale , Zhiwei Sui , Anna Baoutina , Michael Dobeson , Lina Partis , Jayne B. Morrow
{"title":"International interlaboratory study comparing single organism 16S rRNA gene sequencing data: Beyond consensus sequence comparisons","authors":"Nathan D. Olson , Steven P. Lund , Justin M. Zook , Fabiola Rojas-Cornejo , Brian Beck , Carole Foy , Jim Huggett , Alexandra S. Whale , Zhiwei Sui , Anna Baoutina , Michael Dobeson , Lina Partis , Jayne B. Morrow","doi":"10.1016/j.bdq.2015.01.004","DOIUrl":null,"url":null,"abstract":"<div><p>This study presents the results from an interlaboratory sequencing study for which we developed a novel high-resolution method for comparing data from different sequencing platforms for a multi-copy, paralogous gene. The combination of PCR amplification and 16S ribosomal RNA gene (16S rRNA) sequencing has revolutionized bacteriology by enabling rapid identification, frequently without the need for culture. To assess variability between laboratories in sequencing 16S rRNA, six laboratories sequenced the gene encoding the 16S rRNA from <em>Escherichia coli</em> O157:H7 strain EDL933 and <em>Listeria monocytogenes</em> serovar 4b strain NCTC11994. Participants performed sequencing methods and protocols available in their laboratories: Sanger sequencing, Roche 454 pyrosequencing<sup>®</sup>, or Ion Torrent PGM<sup>®</sup>. The sequencing data were evaluated on three levels: (1) identity of biologically conserved position, (2) ratio of 16S rRNA gene copies featuring identified variants, and (3) the collection of variant combinations in a set of 16S rRNA gene copies. The same set of biologically conserved positions was identified for each sequencing method. Analytical methods using Bayesian and maximum likelihood statistics were developed to estimate variant copy ratios, which describe the ratio of nucleotides at each identified biologically variable position, as well as the likely set of variant combinations present in 16S rRNA gene copies. Our results indicate that estimated variant copy ratios at biologically variable positions were only reproducible for high throughput sequencing methods. Furthermore, the likely variant combination set was only reproducible with increased sequencing depth and longer read lengths. We also demonstrate novel methods for evaluating variable positions when comparing multi-copy gene sequence data from multiple laboratories generated using multiple sequencing technologies.</p></div>","PeriodicalId":38073,"journal":{"name":"Biomolecular Detection and Quantification","volume":"3 ","pages":"Pages 17-24"},"PeriodicalIF":0.0000,"publicationDate":"2015-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.bdq.2015.01.004","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomolecular Detection and Quantification","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2214753515000054","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/3/5 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 5
Abstract
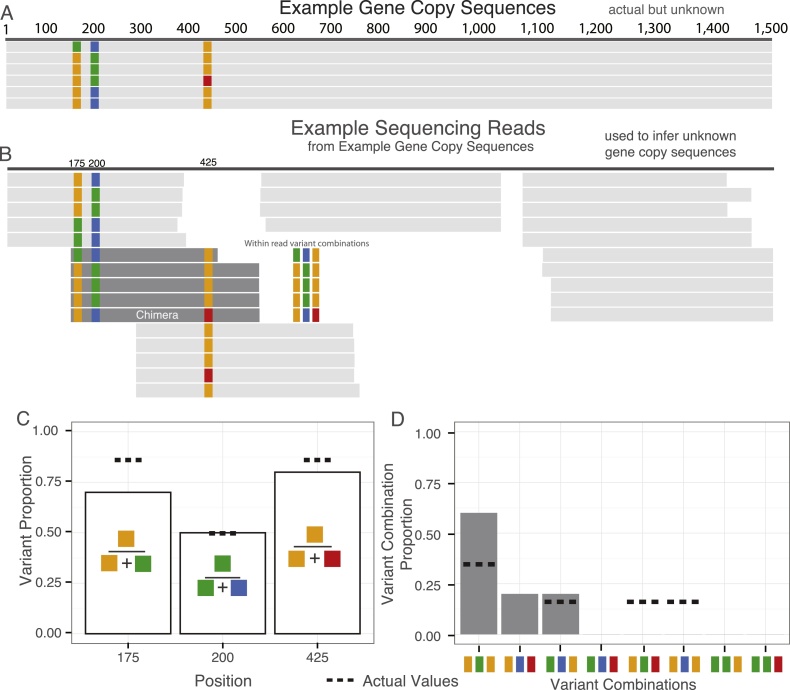
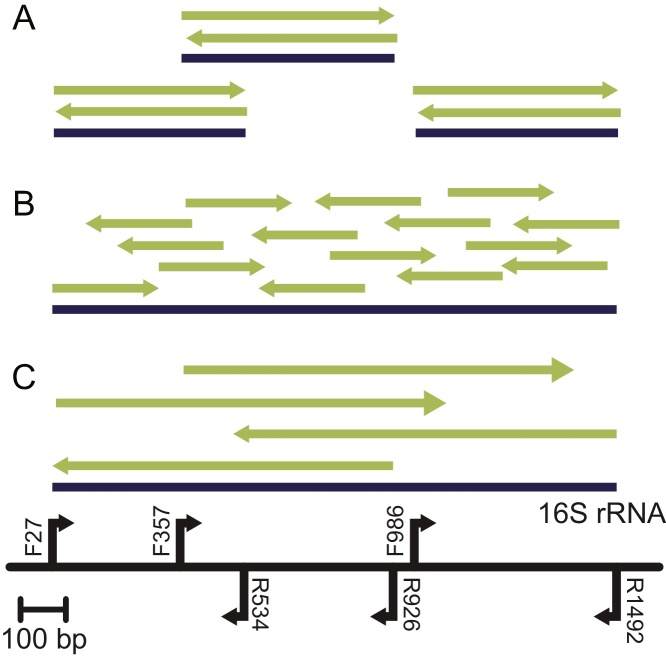
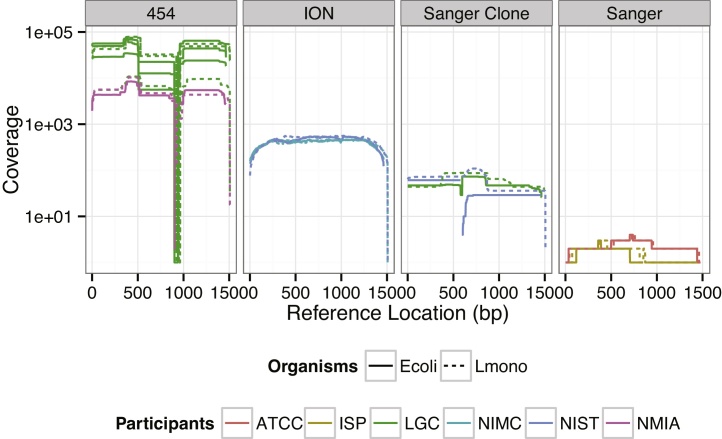
This study presents the results from an interlaboratory sequencing study for which we developed a novel high-resolution method for comparing data from different sequencing platforms for a multi-copy, paralogous gene. The combination of PCR amplification and 16S ribosomal RNA gene (16S rRNA) sequencing has revolutionized bacteriology by enabling rapid identification, frequently without the need for culture. To assess variability between laboratories in sequencing 16S rRNA, six laboratories sequenced the gene encoding the 16S rRNA from Escherichia coli O157:H7 strain EDL933 and Listeria monocytogenes serovar 4b strain NCTC11994. Participants performed sequencing methods and protocols available in their laboratories: Sanger sequencing, Roche 454 pyrosequencing®, or Ion Torrent PGM®. The sequencing data were evaluated on three levels: (1) identity of biologically conserved position, (2) ratio of 16S rRNA gene copies featuring identified variants, and (3) the collection of variant combinations in a set of 16S rRNA gene copies. The same set of biologically conserved positions was identified for each sequencing method. Analytical methods using Bayesian and maximum likelihood statistics were developed to estimate variant copy ratios, which describe the ratio of nucleotides at each identified biologically variable position, as well as the likely set of variant combinations present in 16S rRNA gene copies. Our results indicate that estimated variant copy ratios at biologically variable positions were only reproducible for high throughput sequencing methods. Furthermore, the likely variant combination set was only reproducible with increased sequencing depth and longer read lengths. We also demonstrate novel methods for evaluating variable positions when comparing multi-copy gene sequence data from multiple laboratories generated using multiple sequencing technologies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
